


Pediatric Angioedema without Wheals: How to Guide the Diagnosis
 ,
, 
 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Epidemiology and Pathogenesis
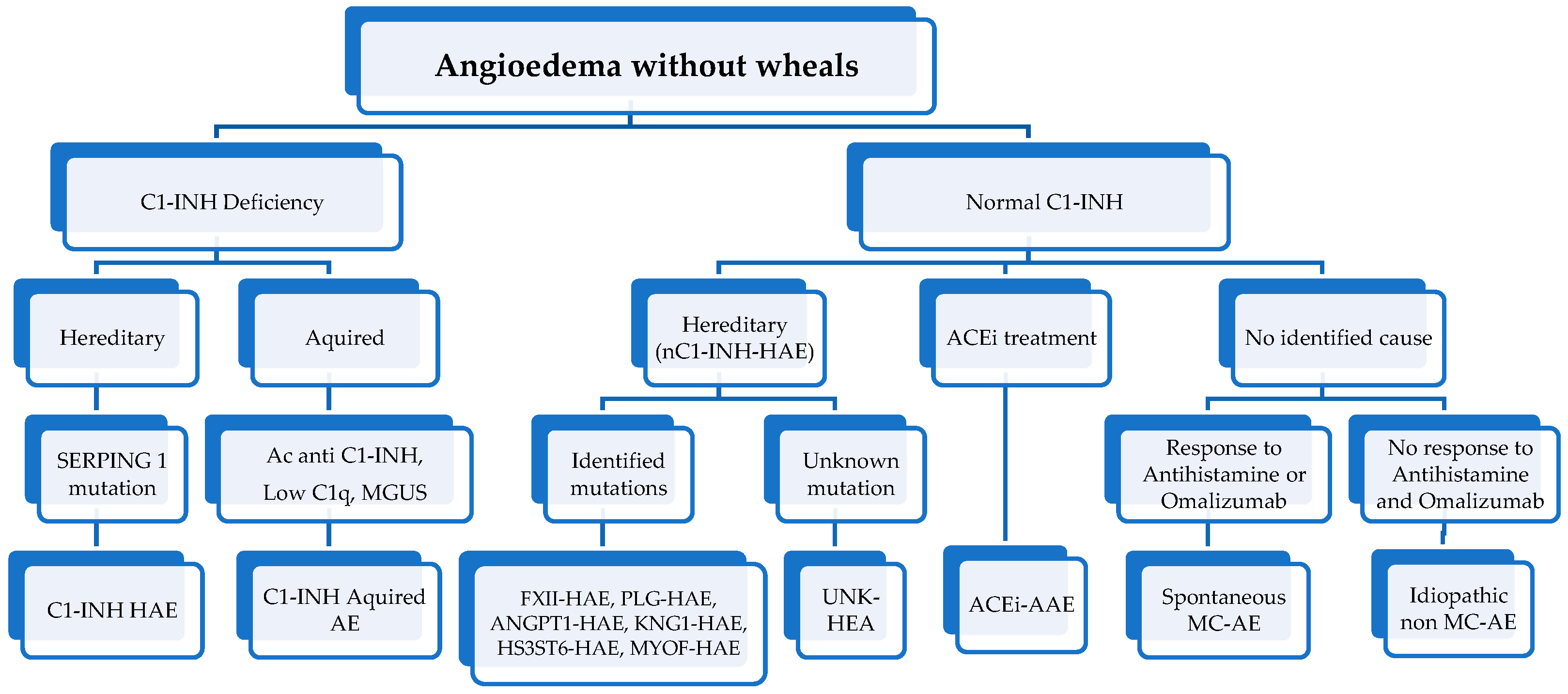
3. Differential Diagnosis
4. Diagnosis
5. Idiopathic AEwW
5.1. Idiopathic Histamine-Mediated AEwW
5.2. Idiopathic Non-Histamine-Mediated AEwW
6. Pediatric Features of Acquired or Idiopathic Histamine-Mediated AEwW
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AE: angioedema |
| AEwW: AE without wheals |
| HAE: hereditary angioedema |
| C1-INH: C1-inhibitor |
| NSAID: Non-steroidal anti-inflammatory drugs |
| VE: vascular endothelial |
| AAE: acquired angioedema |
References
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022, 77, 1961–1990. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Donnelly, J.P.; McAnnally, J.R.; Wang, H.E. National estimates of emergency department visits for angioedema and allergic reactions in the United States. Allergy Asthma Proc. 2013, 34, 150–154. [Google Scholar] [CrossRef]
- Zuberbier, T.; Abdul Latiff, A.H.; Abuzakouk, M.; Aquilina, S.; Asero, R.; Baker, D.; Ballmer-Weber, B.; Bangert, C.; Ben-Shoshan, M.; Bernstein, J.A.; et al. The international EAACI/GA²LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy 2022, 77, 734–766. [Google Scholar] [CrossRef]
- Caffarelli, C.; Paravati, F.; El Hachem, M.; Duse, M.; Bergamini, M.; Simeone, G.; Barbagallo, M.; Bernardini, R.; Bottau, P.; Bugliaro, F.; et al. Management of chronic urticaria in children: A clinical guideline. Ital. J. Pediatr. 2019, 45, 101. [Google Scholar] [CrossRef]
- Pattanaik, D.; Lieberman, J.A. Pediatric Angioedema. Curr. Allergy Asthma Rep. 2017, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.A.; Perego, F.; Zanichelli, A.; Cicardi, M. Angioedema Phenotypes: Disease Expression and Classification. Clin. Rev. Allergy Immunol. 2016, 51, 162–169. [Google Scholar] [CrossRef]
- Sharma, J.; Jindal, A.K.; Banday, A.Z.; Kaur, A.; Rawat, A.; Singh, S.; Longhurst, H. Pathophysiology of Hereditary Angioedema (HAE) Beyond the SERPING1 Gene. Clin. Rev. Allergy Immunol. 2021, 60, 305–315. [Google Scholar] [CrossRef]
- Belbézier, A.; Bocquet, A.; Bouillet, L. Idiopathic Angioedema: Current Challenges. J. Asthma Allergy 2020, 13, 137–144. [Google Scholar] [CrossRef]
- Ertoy Karagol, H.I.; Yilmaz, O.; Bakirtas, A.; Topal, E.; Demirsoy, M.S.; Turktas, I. Angioedema without urticaria in childhood. Pediatr. Allergy Immunol. 2013, 24, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Malbrán, E.; Romero, D.F.; Juri, M.C.; Larrauri, B.J.; Malbrán, A. Epidemiology of angioedema without wheals in an allergy and immunology center. Medicina (Buenos Aires) 2015, 75, 273–276. [Google Scholar]
- Cicardi, M.; Zanichelli, A. Diagnosing angioedema. Immunol. Allergy Clin. North Am. 2013, 33, 449–456. [Google Scholar] [CrossRef]
- Tachdjian, R.; Johnston, D.J. Angioedema: Differential diagnosis and acute management. Postgrad. Med. 2021, 133, 765–770. [Google Scholar] [CrossRef]
- Fok, J.S.; Katelaris, C.H. Angioedema Masqueraders. Clin. Exp. Allergy 2019, 49, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.; Buchheit, K.; Cahill, K.N. Aspirin-exacerbated diseases: Advances in asthma with nasal polyposis, urticaria, angioedema, and anaphylaxis. Curr. Allergy Asthma Rep. 2015, 15, 69. [Google Scholar] [CrossRef]
- Capriles-Behrens, E.; Caplin, J.; Sanchez-Borges, M. NSAID facial angioedema in a selected pediatric atopic population. J. Investig. Allergol. Clin. Immunol. 2000, 10, 277–279. [Google Scholar] [PubMed]
- Sanchez-Borges, M.; Capriles-Behrens, E.; Caballero-Fonseca, F. Hypersensitivity to non-steroidal anti-inflammatory drugs in childhood. Pediatr. Allergy Immunol. 2004, 15, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L. The pathophysiology of hereditary angioedema. World Allergy Organ. J. 2010, 3, S25–S28. [Google Scholar]
- Lang, D.M.; Aberer, W.; Bernstein, J.A.; Chng, H.H.; Grumach, A.; Hide, M.; Maurer, M.; Weber, R.; Zuraw, B. International consensus on hereditary and acquired angioedema. Ann. Allergy Asthma Immunol. 2012, 109, 395–402. [Google Scholar] [CrossRef]
- Fok, J.S.; Katelaris, C.H.; Brown, A.F.; Smith, W.B. Icatibant in angiotensinconverting enzyme (ACE) inhibitor-associated angioedema. Intern. Med. J. 2015, 45, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Zingale, L.C.; Beltrami, L.; Zanichelli, A.; Maggioni, L.; Pappalardo, E.; Cicardi, B.; Cicardi, M. Angioedema without urticaria: A large clinical survey. CMAJ 2006, 175, 1065–1070. [Google Scholar] [CrossRef]
- Agostoni, A.; Aygören-Pürsün, E.; Binkley, K.E.; Blanch, A.; Bork, K.; Bouillet, L.; Bucher, C.; Castaldo, A.J.; Cicardi, M.; Davisiii, A. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J. Allergy Clin. Immunol. 2004, 114, S51–S131. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.L.; Stallings, A.P.; Platts-Mills, T.A.; Oliveira, W.M.; Workman, L.; James, H.R.; Tripathi, A.; Lane, C.J.; Matos, L.; Heymann, P.W.; et al. Galactose-alpha-1,3-galactose and delayed anaphylaxis, angioedema, and urticaria in children. Pediatrics 2013, 131, e1545–e1552. [Google Scholar] [CrossRef] [PubMed]
- Veronez, C.L.; Grumach, A.S. Angioedema without urticaria: Novel findings which must be measured in clinical setting. Curr. Opin. Allergy Clin. Immunol. 2020, 20, 253–260. [Google Scholar] [CrossRef]
- Gleich, G.J.; Schroeter, A.L.; Marcoux, J.P.; Sachs, M.I.; O'Connell, E.J.; Kohler, P.F. Episodic angioedema associated with eosinophilia. N. Engl. J. Med. 1984, 310, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Chikama, R.; Hosokawa, M.; Miyazawa, T.; Miura, R.; Suzuki, T.; Tagami, H. Non episodic angioedema associated with eosinophilia: Report of 4 cases and review of 33 young female patients reported in Japan. Dermatology, 1998; 197, 321–325. [Google Scholar]
- Wehl, G.; Rauchenzauner, M. A Systematic Review of the Literature of the Three Related Disease Entities Cheilitis Granulomatosa, Orofacial Granulomatosis and Melkersson-Rosenthal Syndrome. Curr. Pediatr. Rev. 2018, 14, 196–203. [Google Scholar] [CrossRef]
- Druey, K.M.; Parikh, S.M. Idiopathic systemic capillary leak syndrome (Clarkson disease). J. Allergy Clin. Immunol. 2017, 140, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Naguwa, S.M.; Gershwin, M.E. Pediatric angioedema. Clin. Rev. Allergy Immunol. 2008, 34, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.F.; Khan, D.A.; Brown, E.S.; Bernstein, J.A. Factitious angioedema: A mimic of refractory “angioedema”. J. Allergy Clin. Immunol. Pract. 2014, 2, 795–797. [Google Scholar] [CrossRef]
- Pedrosa, M.; Phillips-Angles, E.; López-Lera, A.; López-Trascasa, M.; Caballero, T. Complement Study Versus CINH Gene Testing for the Diagnosis of Type I Hereditary Angioedema in Children. J. Clin. Immunol. 2016, 36, 16–18. [Google Scholar] [CrossRef]
- Frank, M.M.; Zuraw, B.; Banerji, A.; Bernstein, J.A.; Craig, T.; Busse, P.; Christiansen, S.; Davis-Lorton, M.; Li, H.H.; Lumry, W.R.; et al. Management of children with hereditary angioedema due to C1 inhibitor deficiency. Pediatrics 2016, 138, 5. [Google Scholar] [CrossRef]
- Tai, S.; Mascaro, M.; Goldstein, N.A. Angioedema: A review of 367 episodes presenting to three tertiary care hospitals. Ann. Otol. Rhinol. Laryngol. 2010, 119, 836–841. [Google Scholar] [CrossRef]
- Mansi, M.; Zanichelli, A.; Coerezza, A.; Suffritti, C.; Wu, M.A.; Vacchini, R.; Stieber, C.; Cichon, S.; Cicardi, M. Presentation, diagnosis and treatment of angioedema without wheals: A retrospective analysis of a cohort of 1058 patients. J. Intern. Med. 2015, 277, 585–593. [Google Scholar] [CrossRef]
- van den Elzen, M.; Go, M.F.C.L.; Knulst, A.C.; Blankestijn, M.A.; van Os-Medendorp, H.; Otten, H.G. Efficacy of Treatment of Non-hereditary Angioedema. Clin Rev Allergy Immunol. 2018, 54, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Busse, P.J.; Smith, T. Histaminergic Angioedema. Immunol Allergy Clin. North Am. 2017, 37, 467–481. [Google Scholar] [CrossRef]
- Cicardi, M.; Aberer, W.; Banerji, A.; Bas, M.; Bernstein, J.A.; Bork, K.; Caballero, T.; Farkas, H.; Grumach, A.; Kaplan, A.P.; et al. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the Hereditary Angioedema International Working Group. Allergy 2014, 69, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Wintenberger, C.; Boccon-Gibod, I.; Launay, D.; Fain, O.; Kanny, G.; Jeandel, P.Y.; Martin, L.; Gompel, A.; Bouillet, L. Tranexamic acid as maintenance treatment for non-histaminergic angioedema: Analysis of efficacy and safety in 37 patients. Clin. Exp. Immunol. 2014, 178, 112–117. [Google Scholar] [CrossRef]
- Ocak, M.; Nain, E.; Şahiner, Ü.M.; Akin, M.Ş.; Karabiber, E.; Şekerel, B.E.; Soyer, Ö. Recurrent angioedema in childhood: Hereditary angioedema or histaminergic angioedema? Pediatr. Dermatol. 2021, 38, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Marrouche, N.; Grattan, C. Childhood urticaria. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 485–490. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Types of Angioedema | |||
|---|---|---|---|
| Mediated by Mast Cells | Mediated by Bradykinin | Mediated by Leukotrienes | |
| Pathogenetic Mechanism | • IgE-mediated response to antigen exposure with the release of vasoactive mediators • Non-IgE-mediated (may be associated with chronic spontaneous urticaria) • Idiopathic (unidentifiable cause) | Complex interaction of complement, coagulation, and contact system • Hereditary angioedema with C1-INH deficiency or defect • Hereditary angioedema with normal C1-INH • Acquired angioedema with C1-INH deficiency • ACE inhibitor-induced angioedema | Inhibition of cyclooxygenase-1 determines a shunt of the arachidonic acid metabolism and causes an increase in 5-lipoxygenase activity • Induced by NSAIDs, aspirin |
| Response to antihistamine in 12 h | Yes | No | |
| Urticaria | Frequent | Absent | |
| Age of onset | Anyway | Often in the 1st or 2nd decade (40% within 5 years) | Anyway |
| Pruritus | Present | Slight | |
| Duration of edema | Usually <48 h | Often >72 h | Variable |
| Preferred localizations | Face (eyelids, lips), neck | Face, abdomen, extremities | Periorbital, airways |
| Prodromal symptoms | No | Often | |
| Trauma as a trigger | No | Yes | |
| Development of signs and symptoms | Fast | Slower | Hours |
| Familiar history | Never | Often | Not known |
| Further investigations | Levels of serum tryptase (useful in the context of anaphylaxis/allergenic trigger) | Serum C4 Quantitative and qualitative levels of C1-INH (all normal in ACE inhibitor-induced angioedema) | |
| Demographic, Personal History and Clinical Data | |
|---|---|
| Gender | Male |
| Average age at first-episode onset | 7–7.8 yrs |
| Average age at diagnosis | 8–9 yrs |
| Average length of relapse episode | 24 h |
| Number of exacerbation episodes in the last year | 2–5 |
| Most involved body areas | Eyelids, lips |
| Number of body areas involved during exacerbation episodes | Several |
| Personal history | Atopy |
| Familial history | Absence of family history related to recurrent AE episodes |
| Trigger factors | Infections, respiratory or food allergens, thyroid diseases, drugs (medical history of trauma is not frequent) |
| Response to antihistamine treatment | Symptoms improvement |
| Need for adrenaline or intubation | No |
| Prognosis | Good |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liotti, L.; Pecoraro, L.; Mastrorilli, C.; Castagnoli, R.; Saretta, F.; Mori, F.; Arasi, S.; Barni, S.; Giovannini, M.; Caminiti, L.; et al. Pediatric Angioedema without Wheals: How to Guide the Diagnosis. Life 2023, 13, 1021. https://doi.org/10.3390/life13041021
Liotti L, Pecoraro L, Mastrorilli C, Castagnoli R, Saretta F, Mori F, Arasi S, Barni S, Giovannini M, Caminiti L, et al. Pediatric Angioedema without Wheals: How to Guide the Diagnosis. Life. 2023; 13(4):1021. https://doi.org/10.3390/life13041021
Chicago/Turabian StyleLiotti, Lucia, Luca Pecoraro, Carla Mastrorilli, Riccardo Castagnoli, Francesca Saretta, Francesca Mori, Stefania Arasi, Simona Barni, Mattia Giovannini, Lucia Caminiti, and et al. 2023. "Pediatric Angioedema without Wheals: How to Guide the Diagnosis" Life 13, no. 4: 1021. https://doi.org/10.3390/life13041021
APA StyleLiotti, L., Pecoraro, L., Mastrorilli, C., Castagnoli, R., Saretta, F., Mori, F., Arasi, S., Barni, S., Giovannini, M., Caminiti, L., Miraglia Del Giudice, M., & Novembre, E. (2023). Pediatric Angioedema without Wheals: How to Guide the Diagnosis. Life, 13(4), 1021. https://doi.org/10.3390/life13041021