




Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease

 ,
,  ,
,  ,
, 
Abstract
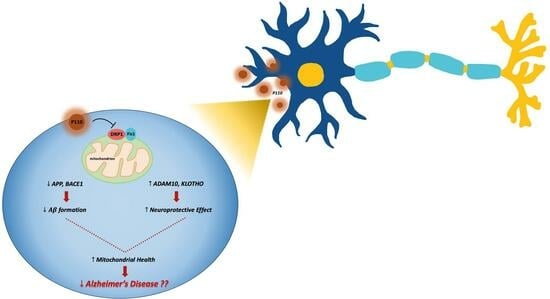
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and P110 Treatment
2.2. Real-Time PCR
2.3. Western Blotting
2.4. Mitotracker Staining
2.5. Measurement of Intracellular Reactive Oxygen Species
2.6. Detection of Mitochondrial ROS Production
2.7. Electron Microscopy
2.8. Statistics
3. Results
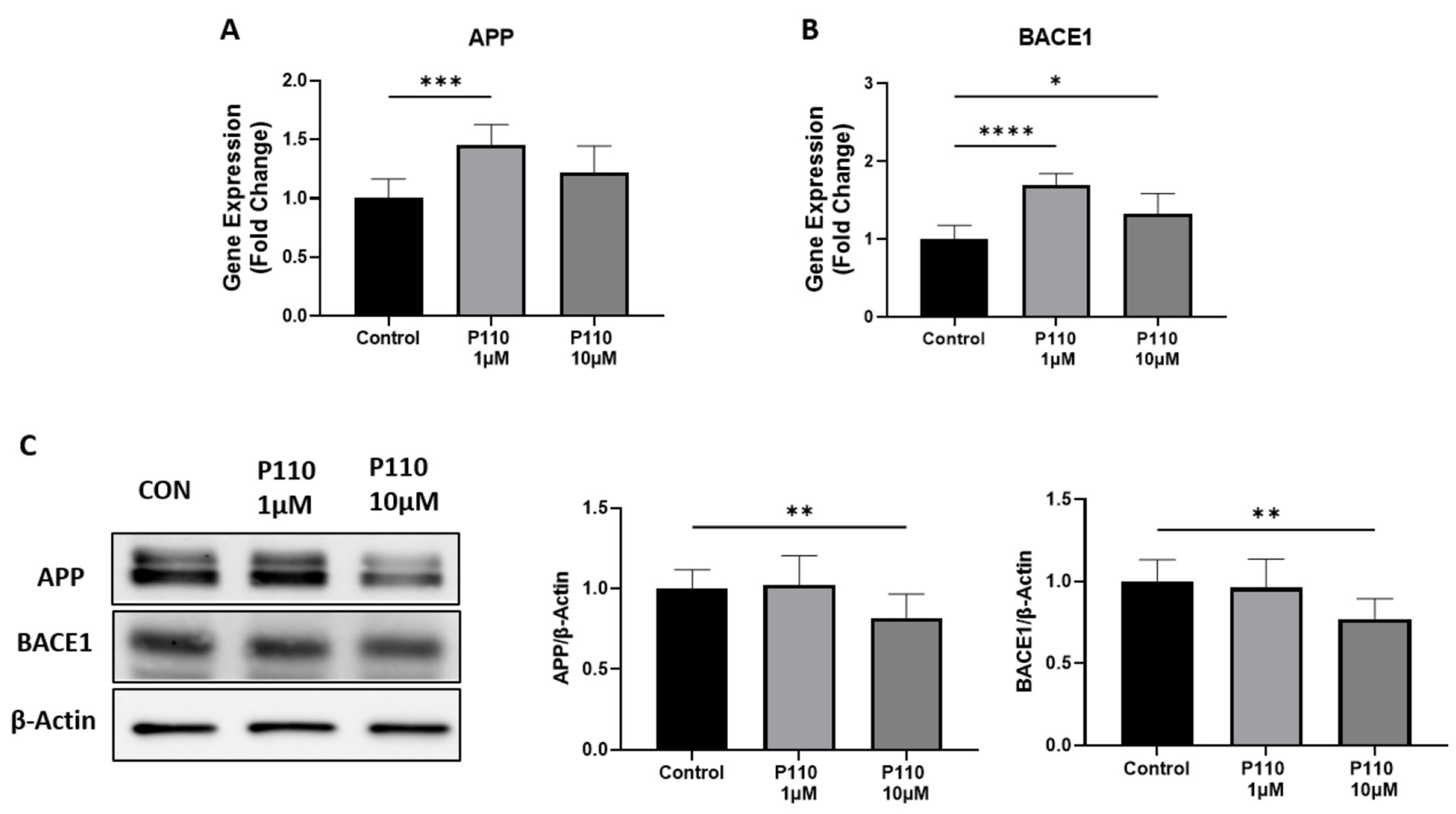
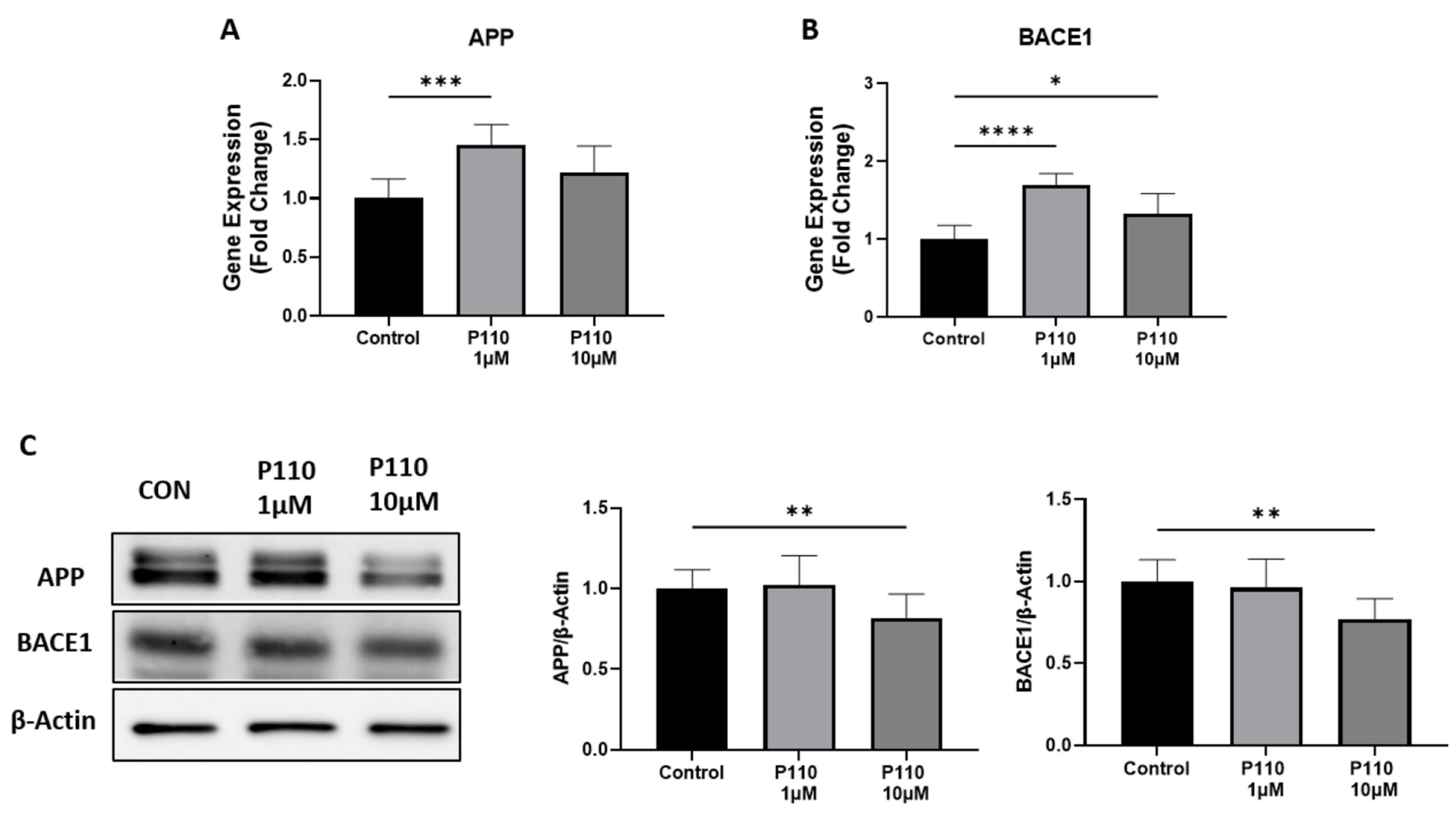
3.1. P110 Treatment Regulates Aβ Formation in SH-SY5Y Cells
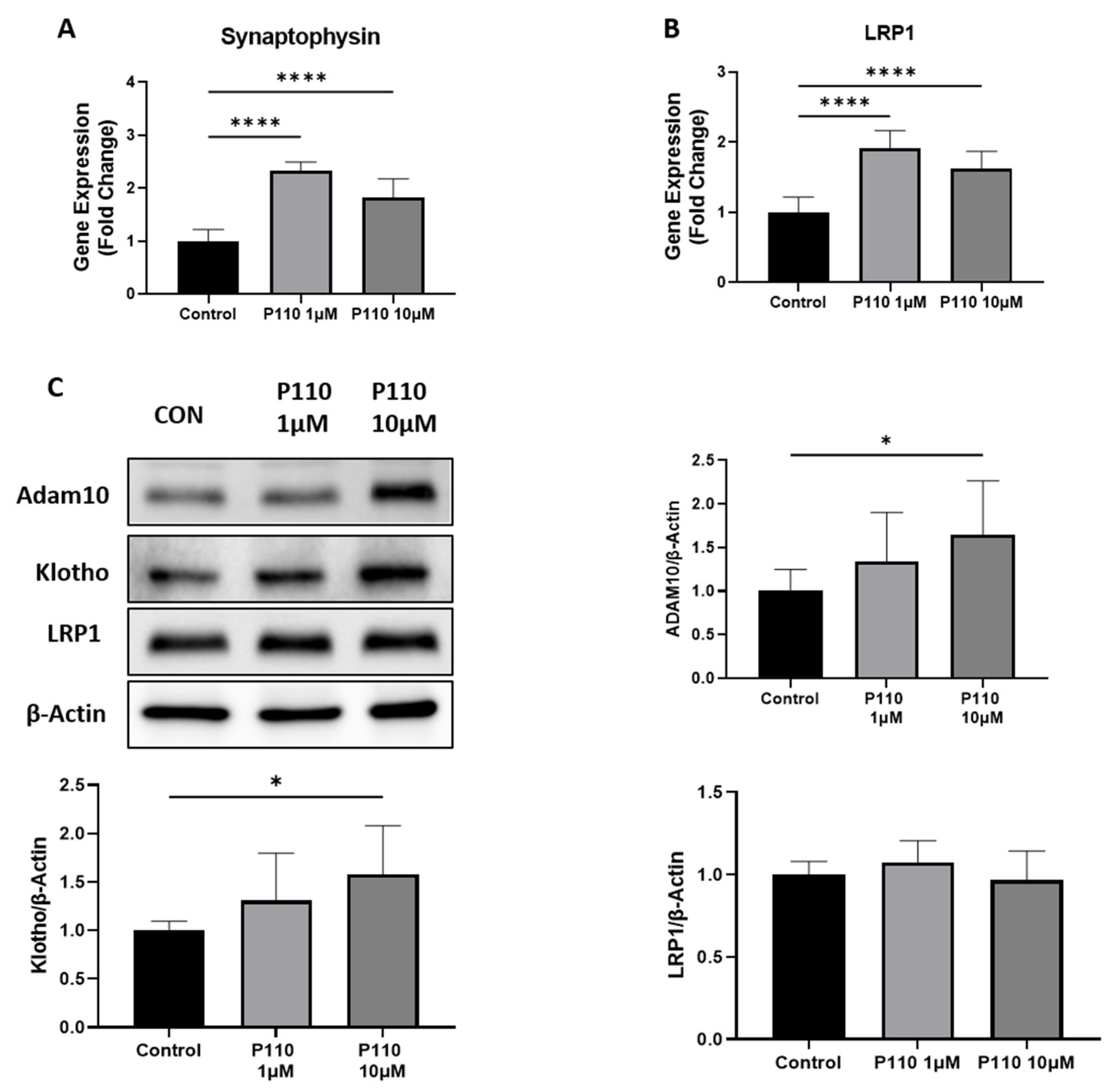
3.2. P110 Treatment Enhances Neuroprotective Effects in SH-SY5Y Cells
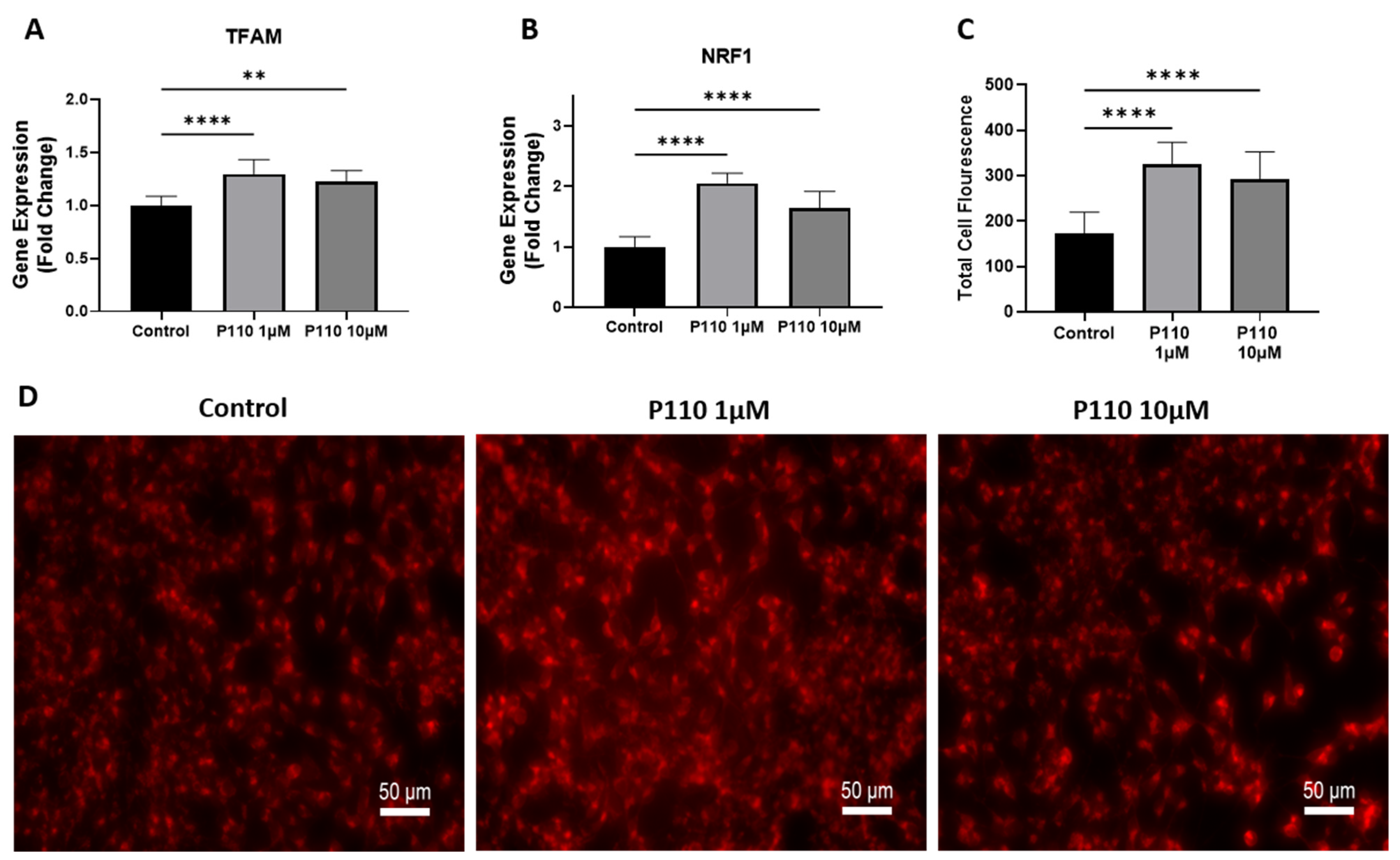
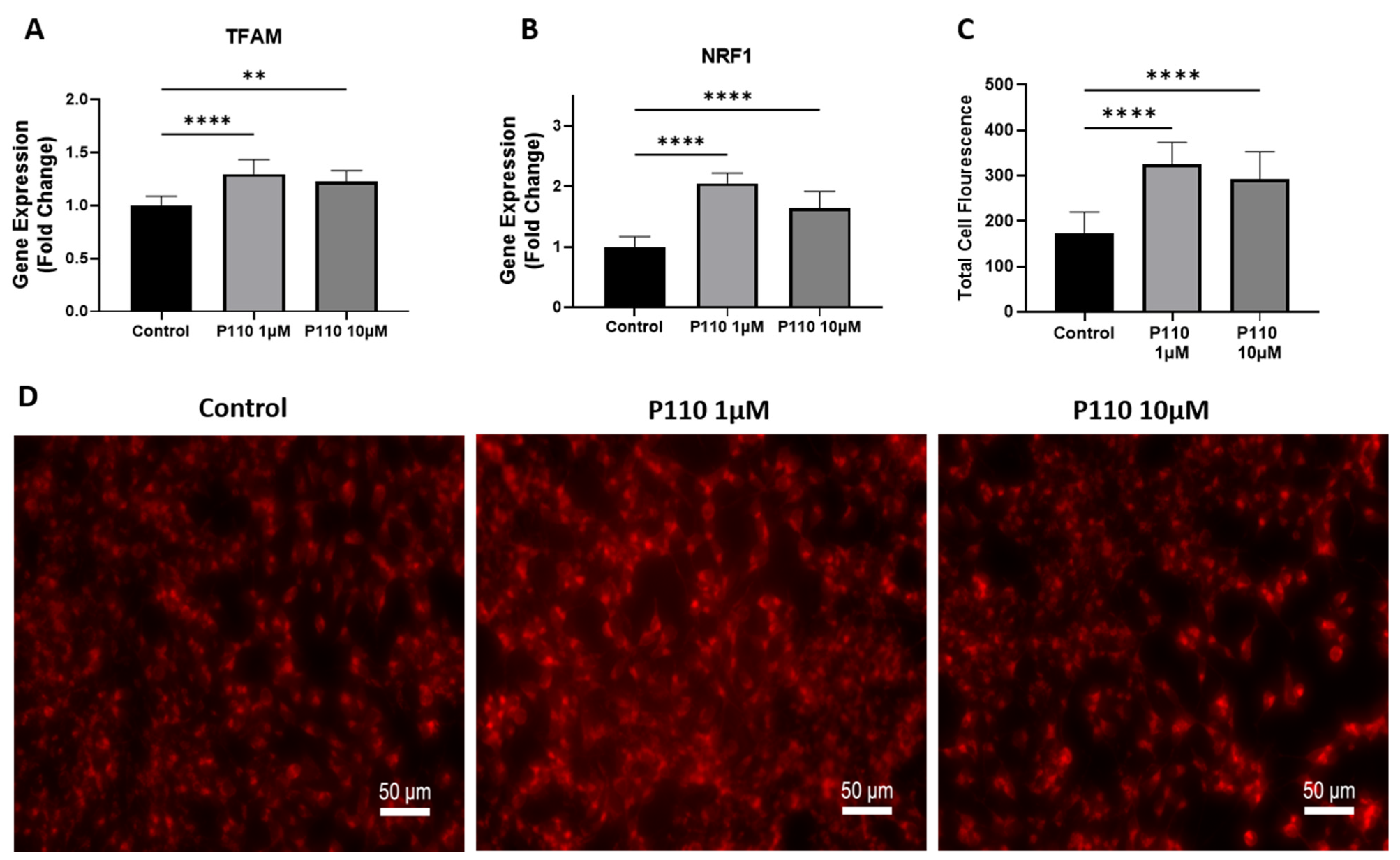
3.3. P110 Treatment Increases Mitochondrial Function
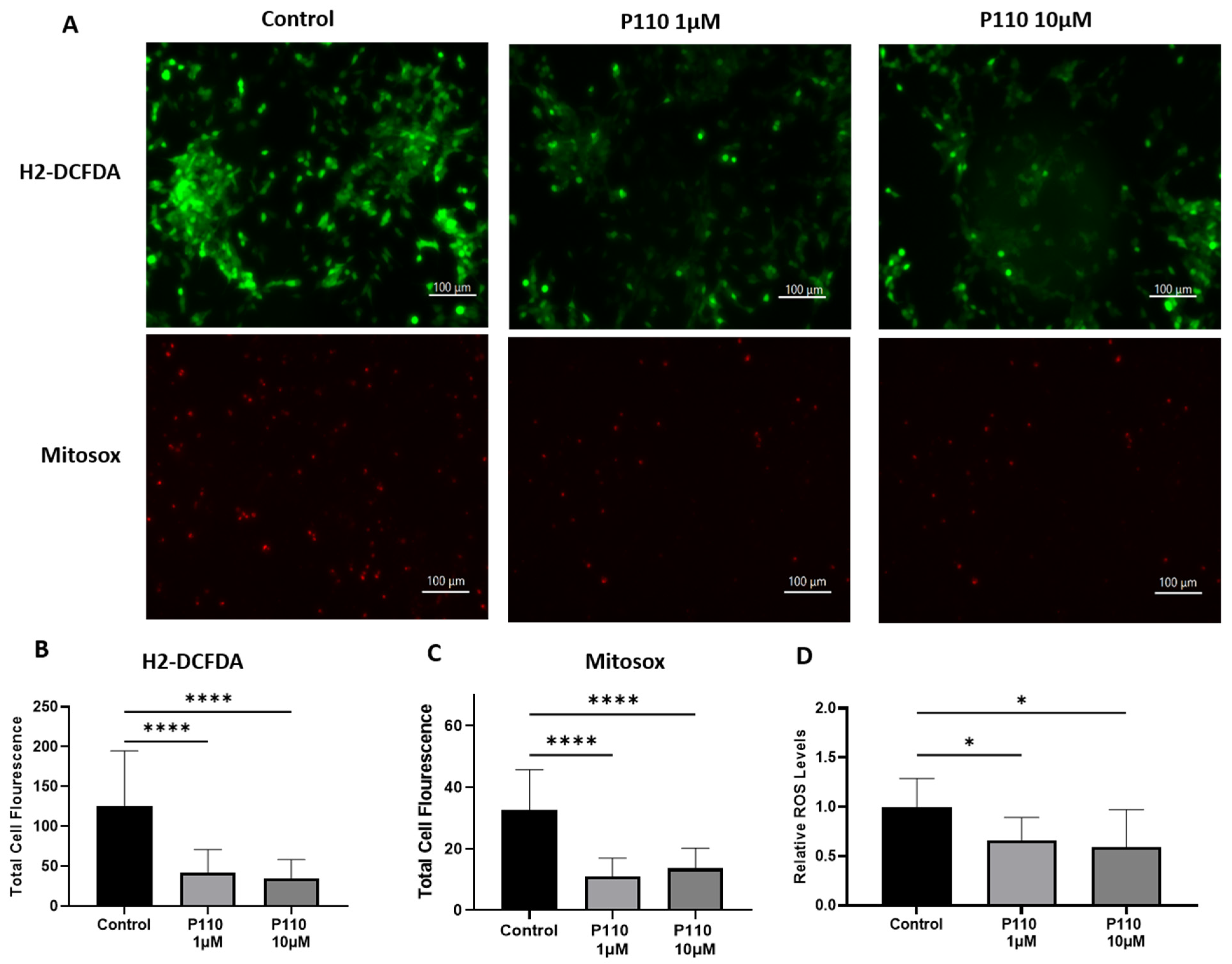
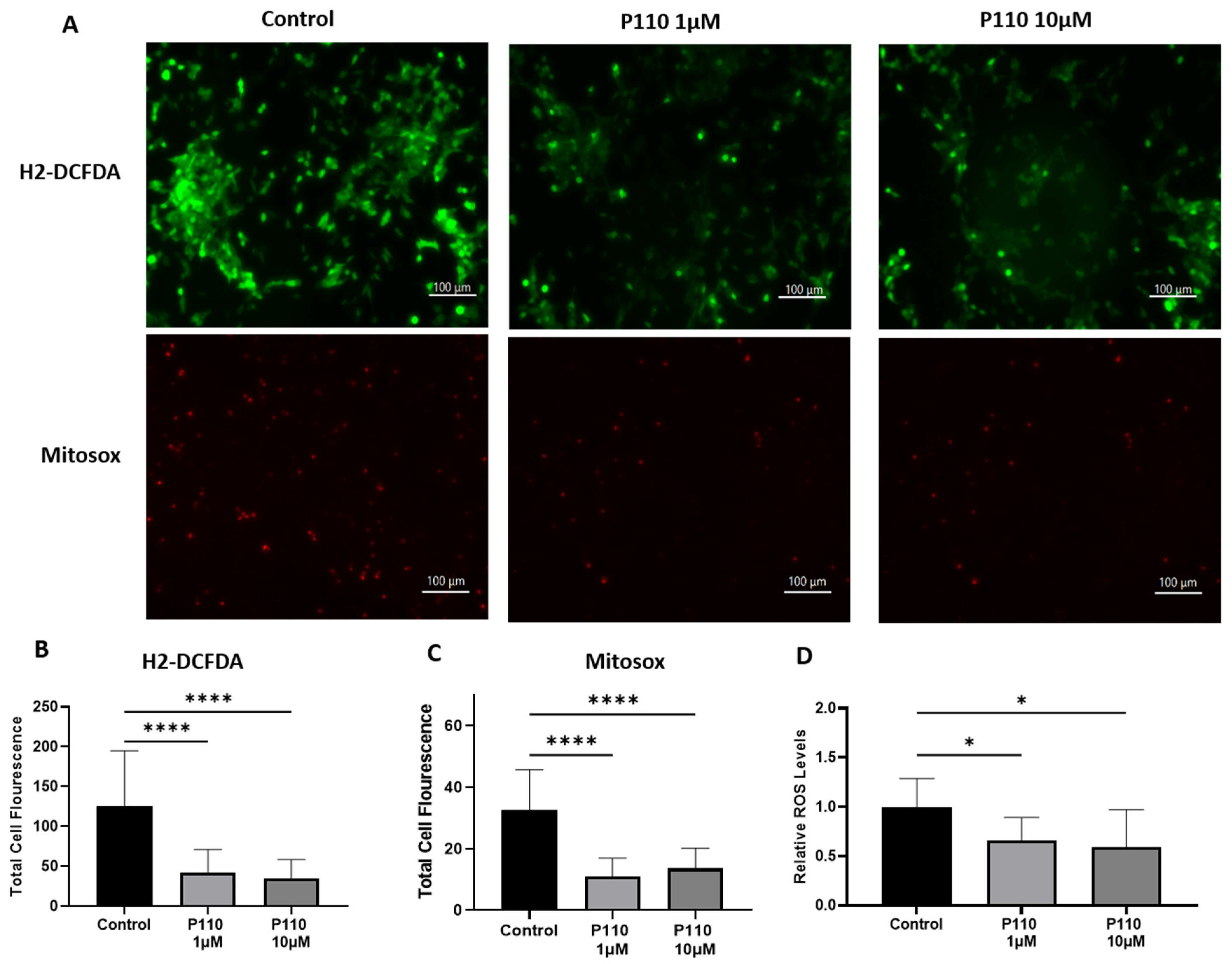
3.4. P110 Treatment Reduces ROS Production
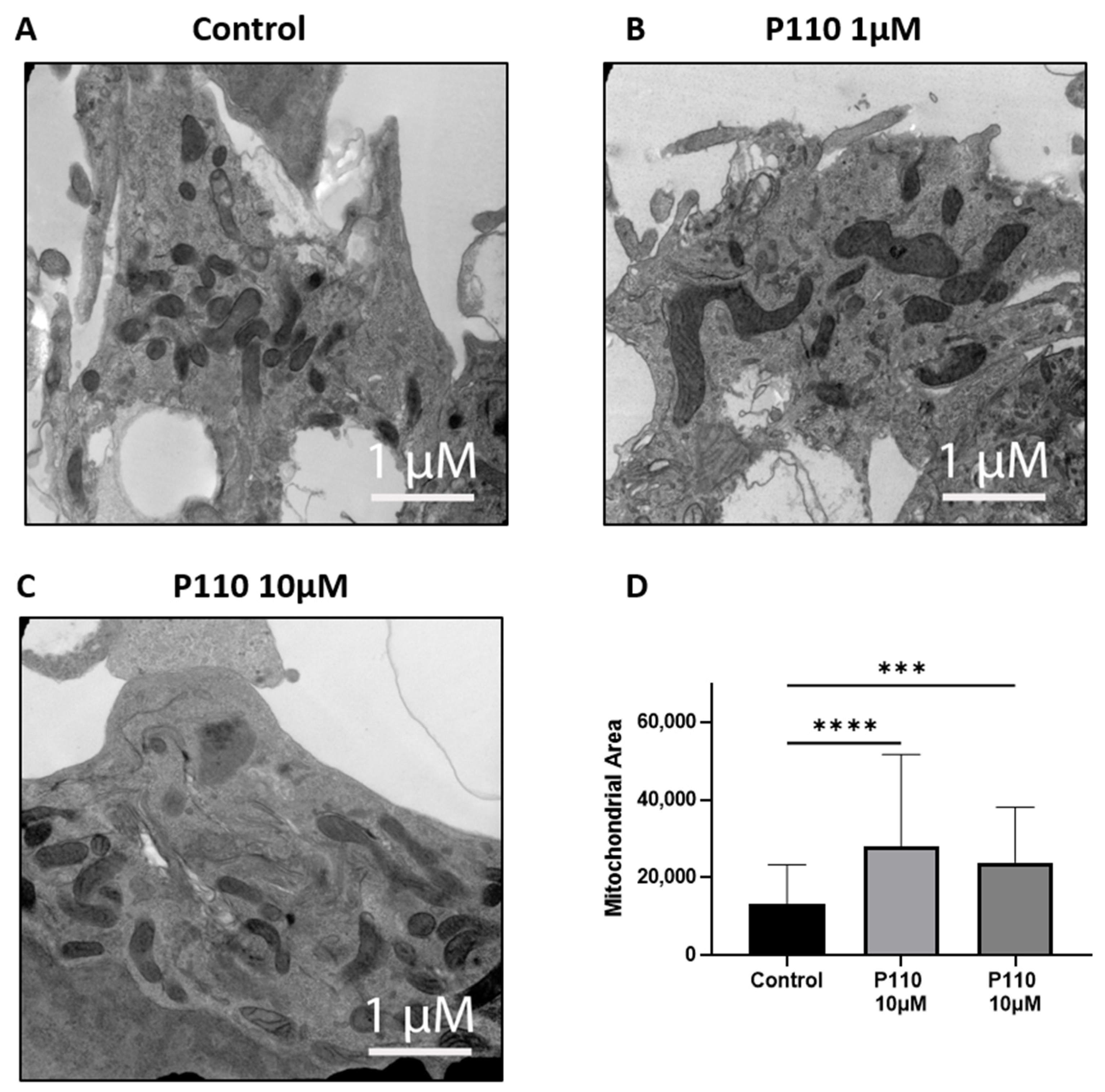
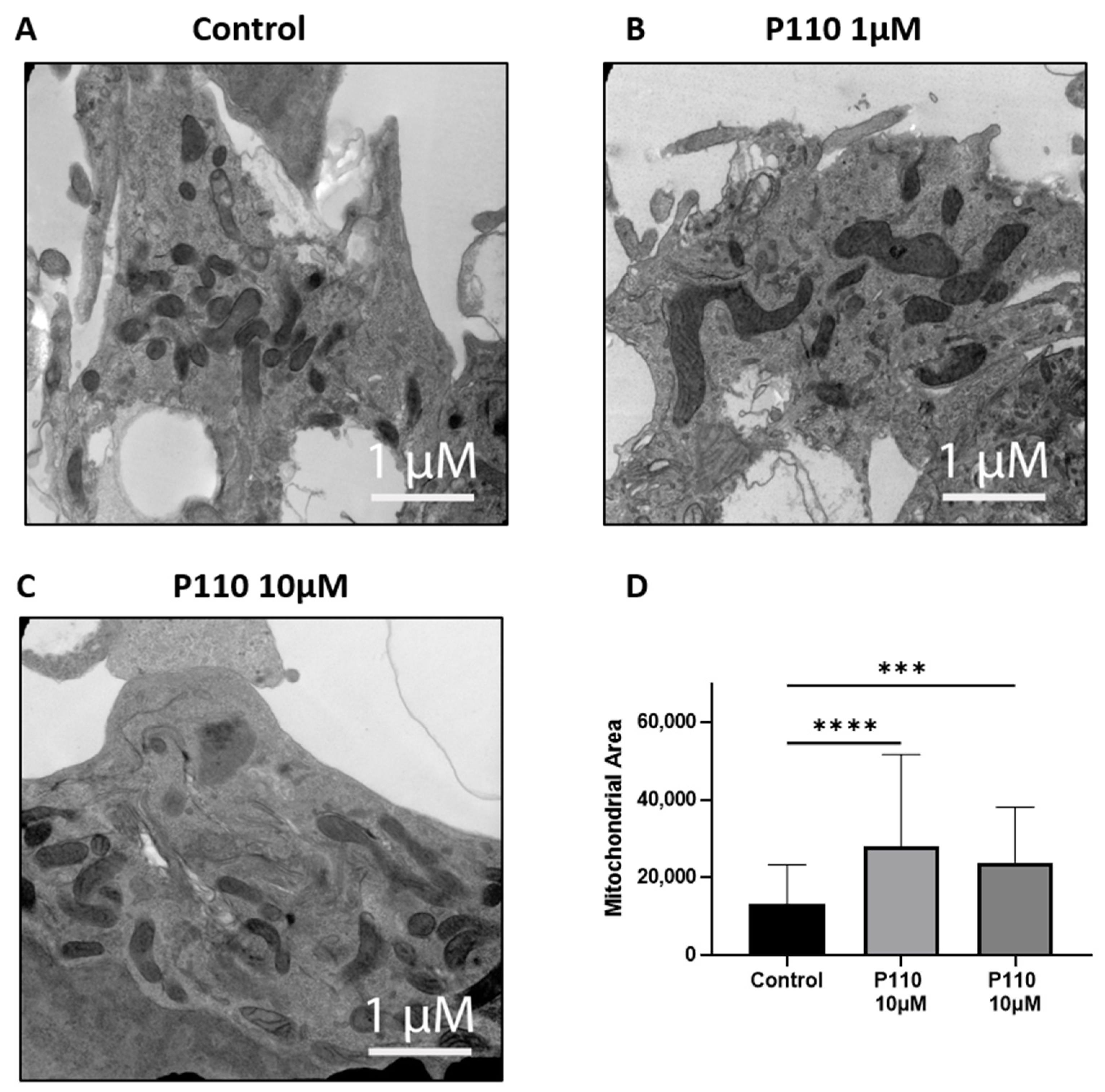
3.5. P110 Treatment Reduces Mitochondrial Fragmentation
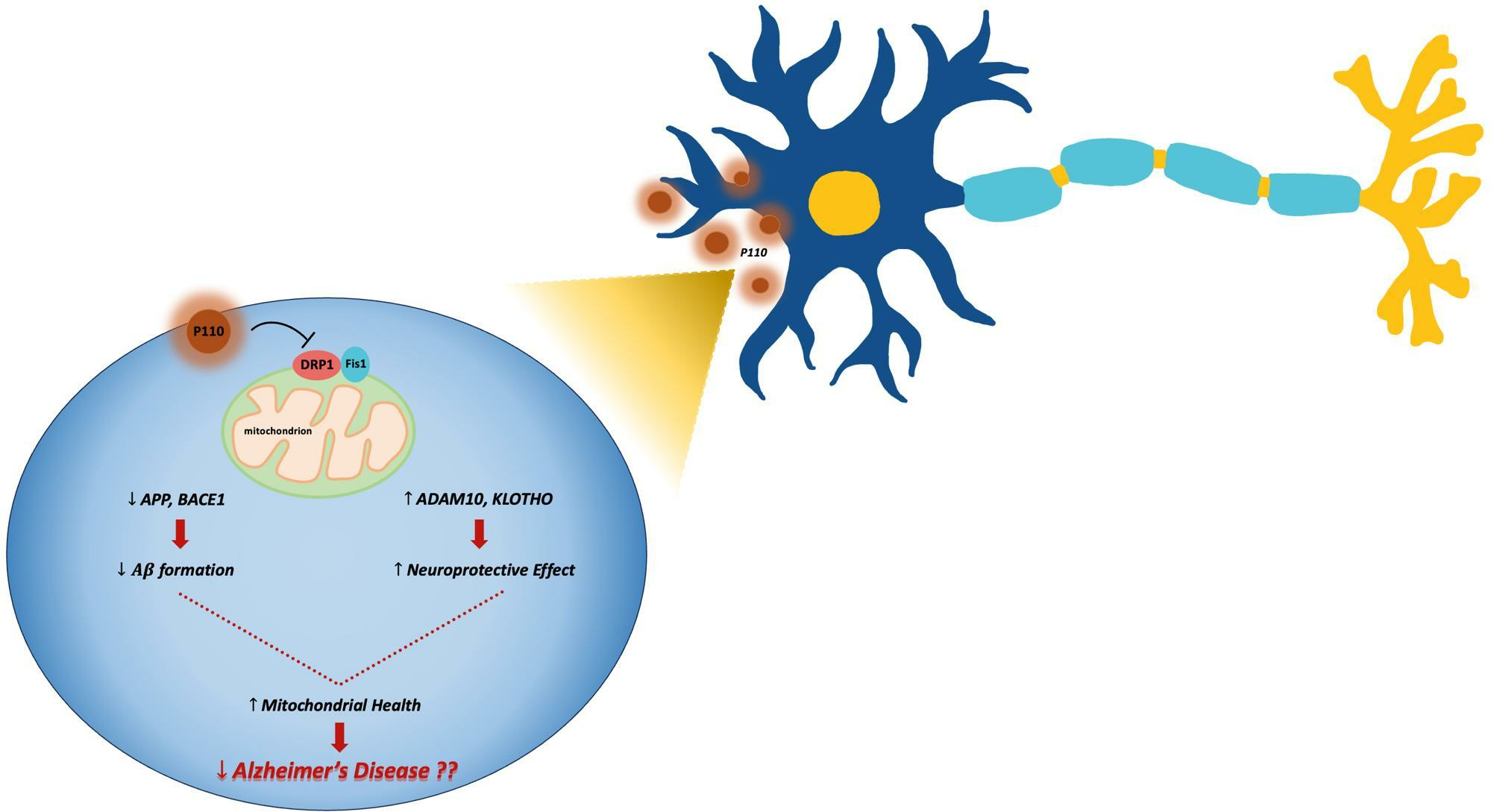
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A.; Emamzadeh, F.N.; Titova, M.; Surguchev, A.A. Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era. Biomedicines 2023, 11, 1215. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef]
- Reiss, A.B.; Muhieddine, D.; Jacob, B.; Mesbah, M.; Pinkhasov, A.; Gomolin, I.H.; Stecker, M.M.; Wisniewski, T.; De Leon, J. Alzheimer’s Disease Treatment: The Search for a Breakthrough. Medicina 2023, 59, 1084. [Google Scholar] [CrossRef] [PubMed]
- Kruk, J.; Sachdev, P.; Singh, S. Neuroleptic-induced respiratory dyskinesia. J. Neuropsychiatry Clin. Neurosci. 1995, 7, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; He, Z.; You, G.; Liu, Q.; Li, N. The Deficits of Insulin Signal in Alzheimer’s Disease and the Mechanisms of Vanadium Compounds in Curing AD. Curr. Issues Mol. Biol. 2023, 45, 6365–6382. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Holubiec, M.I.; Alloatti, M.; Bianchelli, J.; Greloni, F.; Arnaiz, C.; Gonzalez Prinz, M.; Fernandez Bessone, I.; Pozo Devoto, V.; Falzone, T.L. Mitochondrial vulnerability to oxidation in human brain organoids modelling Alzheimer’s disease. Free. Radic. Biol. Med. 2023, 208, 394–401. [Google Scholar] [CrossRef]
- Haigh, J.L.; New, L.E.; Filippi, B.M. Mitochondrial Dynamics in the Brain Are Associated With Feeding, Glucose Homeostasis, and Whole-Body Metabolism. Front. Endocrinol. 2020, 11, 580879. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Perez-Gonzalez, M.; Alfaro-Ruiz, R.; Badesso, S.; Sucunza, D.; Espelosin, M.; Ursua, S.; Lachen-Montes, M.; Fernandez-Irigoyen, J.; Santamaria, E.; et al. Amyloid-Driven Tau Accumulation on Mitochondria Potentially Leads to Cognitive Deterioration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11950. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828. [Google Scholar] [CrossRef] [PubMed]
- Agholme, L.; Lindstrom, T.; Kagedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimer’s Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Mochly-Rosen, D. Mortal engines: Mitochondrial bioenergetics and dysfunction in neurodegenerative diseases. Pharmacol. Res. 2018, 138, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef]
- Zerihun, M.; Sukumaran, S.; Qvit, N. The Drp1-Mediated Mitochondrial Fission Protein Interactome as an Emerging Core Player in Mitochondrial Dynamics and Cardiovascular Disease Therapy. Int. J. Mol. Sci. 2023, 24, 5785. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
- Lardelli, M. An Alternative View of Familial Alzheimer’s Disease Genetics. J. Alzheimer’s Dis. 2023, 96, 13–39. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Tacal, O. Trafficking and proteolytic processing of amyloid precursor protein and secretases in Alzheimer’s disease development: An up-to-date review. Eur. J. Pharmacol. 2019, 856, 172415. [Google Scholar] [CrossRef]
- Hershkovits, A.S.; Gelley, S.; Hanna, R.; Kleifeld, O.; Shulman, A.; Fishman, A. Shifting the balance: Soluble ADAM10 as a potential treatment for Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1171123. [Google Scholar] [CrossRef]
- Fung, T.Y.; Iyaswamy, A.; Sreenivasmurthy, S.G.; Krishnamoorthi, S.; Guan, X.J.; Zhu, Z.; Su, C.F.; Liu, J.; Kan, Y.; Zhang, Y.; et al. Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review. Biomedicines 2022, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Olive Biophenols Reduces Alzheimer’s Pathology in SH-SY5Y Cells and APPswe Mice. Int. J. Mol. Sci. 2018, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, I.; Norlin, M. Protective effects of 27- and 24-hydroxycholesterol against staurosporine-induced cell death in undifferentiated neuroblastoma SH-SY5Y cells. Neurosci. Lett. 2012, 525, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.K.; Donohue, K.B.; Gust, K.A. Cellular and molecular responses to ethyl-parathion in undifferentiated SH-SY5Y cells provide neurotoxicity pathway indicators for organophosphorus impacts. Toxicol. Sci. 2023, 191, 285–295. [Google Scholar] [CrossRef]
- Zhou, X.; Bouitbir, J.; Liechti, M.E.; Krahenbuhl, S.; Mancuso, R.V. Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 2841. [Google Scholar] [CrossRef]
- Cheng, N.; Jiao, S.; Gumaste, A.; Bai, L.; Belluscio, L. APP Overexpression Causes Abeta-Independent Neuronal Death through Intrinsic Apoptosis Pathway. eNeuro 2016, 3, ENEURO.0150-16.2016. [Google Scholar] [CrossRef]
- Carriba, P.; Jimenez, S.; Navarro, V.; Moreno-Gonzalez, I.; Barneda-Zahonero, B.; Moubarak, R.S.; Lopez-Soriano, J.; Gutierrez, A.; Vitorica, J.; Comella, J.X. Amyloid-beta reduces the expression of neuronal FAIM-L, thereby shifting the inflammatory response mediated by TNFalpha from neuronal protection to death. Cell Death Dis. 2015, 6, e1639. [Google Scholar] [CrossRef] [PubMed]
- Rudnitskaya, E.A.; Muraleva, N.A.; Maksimova, K.Y.; Kiseleva, E.; Kolosova, N.G.; Stefanova, N.A. Melatonin Attenuates Memory Impairment, Amyloid-beta Accumulation, and Neurodegeneration in a Rat Model of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 103–116. [Google Scholar] [CrossRef]
- Stockley, J.H.; Ravid, R.; O’Neill, C. Altered beta-secretase enzyme kinetics and levels of both BACE1 and BACE2 in the Alzheimer’s disease brain. FEBS Lett. 2006, 580, 6550–6560. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- McDade, E.; Voytyuk, I.; Aisen, P.; Bateman, R.J.; Carrillo, M.C.; De Strooper, B.; Haass, C.; Reiman, E.M.; Sperling, R.; Tariot, P.N.; et al. The case for low-level BACE1 inhibition for the prevention of Alzheimer disease. Nat. Rev. Neurol. 2021, 17, 703–714. [Google Scholar] [CrossRef]
- Li, J.; Wang, H. miR-15b reduces amyloid-beta accumulation in SH-SY5Y cell line through targetting NF-kappaB signaling and BACE1. Biosci. Rep. 2018, 38, BSR20180051. [Google Scholar] [CrossRef]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC Genom. 2017, 18, 305. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Jiang, D.; Cope, A.L.; Zhang, J.; Pennell, M. On the Decoupling of Evolutionary Changes in mRNA and Protein Levels. Mol. Biol. Evol. 2023, 40, msad169. [Google Scholar] [CrossRef]
- Ananya, F.N.; Ahammed, M.R.; Lahori, S.; Parikh, C.; Lawrence, J.A.; Sulachni, F.; Barqawi, T.; Kamwal, C. Neuroprotective Role of Klotho on Dementia. Cureus 2023, 15, e40043. [Google Scholar] [CrossRef] [PubMed]
- Torbus-Paluszczak, M.; Bartman, W.; Adamczyk-Sowa, M. Klotho protein in neurodegenerative disorders. Neurol. Sci. 2018, 39, 1677–1682. [Google Scholar] [CrossRef]
- Kuang, X.; Chen, Y.S.; Wang, L.F.; Li, Y.J.; Liu, K.; Zhang, M.X.; Li, L.J.; Chen, C.; He, Q.; Wang, Y.; et al. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 169–178. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res. Mol. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2002, 28, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Escobar-Khondiker, M.; Hollerhage, M.; Muriel, M.P.; Champy, P.; Bach, A.; Depienne, C.; Respondek, G.; Yamada, E.S.; Lannuzel, A.; Yagi, T.; et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. 2007, 27, 7827–7837. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef]
- Hilt, S.; Altman, R.; Kalai, T.; Maezawa, I.; Gong, Q.; Wachsmann-Hogiu, S.; Jin, L.W.; Voss, J.C. A Bifunctional Anti-Amyloid Blocks Oxidative Stress and the Accumulation of Intraneuronal Amyloid-Beta. Molecules 2018, 23, 2010. [Google Scholar] [CrossRef]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e586. [Google Scholar] [CrossRef]
- Sedlack, A.J.H.; Saleh-Anaraki, K.; Kumar, S.; Ear, P.H.; Lines, K.E.; Roper, N.; Pacak, K.; Bergsland, E.; Quelle, D.E.; Howe, J.R.; et al. Preclinical Models of Neuroendocrine Neoplasia. Cancers 2022, 14, 5646. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Yang, Y.; Tang, H.; Si, Y.; Chen, Z.; Shi, Y.; Fang, H. Metformin Protects Against Diabetes-Induced Cognitive Dysfunction by Inhibiting Mitochondrial Fission Protein DRP1. Front. Pharmacol. 2022, 13, 832707. [Google Scholar] [CrossRef]
- Park, S.J.; Bae, J.E.; Jo, D.S.; Kim, J.B.; Park, N.Y.; Fang, J.; Jung, Y.K.; Jo, D.G.; Cho, D.H. Increased O-GlcNAcylation of Drp1 by amyloid-beta promotes mitochondrial fission and dysfunction in neuronal cells. Mol. Brain 2021, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Zhu, Y.; Zhang, X.L.; Wang, L.; Zhou, Y.M.; Zhang, F.F.; Zhang, H.T.; Zhao, X.M. Baicalin attenuates amyloid beta oligomers induced memory deficits and mitochondria fragmentation through regulation of PDE-PKA-Drp1 signalling. Psychopharmacology 2022, 239, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yang, P.; Yang, Z.J.; Li, Q.G.; Ouyang, Y.T.; Yu, T.; Shangguan, J.H.; Wan, Y.Y.; Jiang, L.P.; Qu, X.H.; et al. Blockage of Drp1 phosphorylation at Ser579 protects neurons against Abeta(1-42)-induced degeneration. Mol. Med. Rep. 2021, 24, 657. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer (Tm) | Forward Sequence | Reverse Sequence |
|---|---|---|
| GAPDH (62 °C) | ACCATCATCCCTGCCTCTAC | CCTGTTGCTGTAGCCAAAT |
| APP (62 °C) | TTTGGCACTGCTCCTGCT | CCACAGAACATGGCAATC |
| BACE-1 (62 °C) | GCAGGGCTACTACGTGGAGA | CAGCACCCACTGCAAAGTTA |
| TFAM (63 °C) | AAGATTCCAAGAAGCTAAGGGTGA | CAGAGTCAGACAGATTTTTTCCAGTTT |
| SYNAPTOPHYSIN (65 °C) | CTGCAATGGGTCTTCGCCA | ACTCTCGGTCTTGTTGGC |
| LRP-1 (63 °C) | ATGGGCAGATCCCAAAGGTG | CAGTCATTGTCATTGTCGCATCT |
| NRF-1 (63 °C) | GGCACTGTCTCACTTATCCAGGTT | CAGCCACGGCAGAATAATTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, A.; Johnson, M.; Renna, H.A.; Sheehan, K.M.; Ahmed, S.; Palaia, T.; Pinkhasov, A.; Gomolin, I.H.; De Leon, J.; Reiss, A.B. Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease. Life 2023, 13, 2156. https://doi.org/10.3390/life13112156
Srivastava A, Johnson M, Renna HA, Sheehan KM, Ahmed S, Palaia T, Pinkhasov A, Gomolin IH, De Leon J, Reiss AB. Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease. Life. 2023; 13(11):2156. https://doi.org/10.3390/life13112156
Chicago/Turabian StyleSrivastava, Ankita, Maryann Johnson, Heather A. Renna, Katie M. Sheehan, Saba Ahmed, Thomas Palaia, Aaron Pinkhasov, Irving H. Gomolin, Joshua De Leon, and Allison B. Reiss. 2023. "Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease" Life 13, no. 11: 2156. https://doi.org/10.3390/life13112156
APA StyleSrivastava, A., Johnson, M., Renna, H. A., Sheehan, K. M., Ahmed, S., Palaia, T., Pinkhasov, A., Gomolin, I. H., De Leon, J., & Reiss, A. B. (2023). Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease. Life, 13(11), 2156. https://doi.org/10.3390/life13112156