
Crosstalk between Glycogen-Selective Autophagy, Autophagy and Apoptosis as a Road towards Modifier Gene Discovery and New Therapeutic Strategies for Glycogen Storage Diseases
 ,
,
Abstract
:1. Introduction
2. Glycogen-Selective Autophagy
2.1. Signal Pathways Involved in Glycogen-Selective Autophagy
2.2. Glycogen-Selective Autophagy and Glycogen Storage Disease I
2.3. Genes Involved in Glycogen-Selective Autophagy
2.4. Other Genes Involved in Autophagy
3. Cross-Talk between Autophagy and Apoptosis
4. Road towards Modifier Gene Discovery
4.1. Rare vs. Common Variants
4.2. Regulatory Role of Non-Coding Regions
4.2.1. Transcription Factors
4.2.2. Non-Coding RNA
5. Therapeutic Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, E.; Andreu, A.L.; Bruno, C. Neuromuscular Disorders of Glycogen Metabolism. Curr. Neurol. Neurosci. Rep. 2013, 13, 333. [Google Scholar] [CrossRef]
- Özen, H. Glycogen storage diseases: New perspectives. World J. Gastroenterol. 2007, 13, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Beckemeyer, A.A. New therapeutic approaches for Pompe disease: Enzyme replacement therapy and beyond. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. S1), 114–124. [Google Scholar]
- Kakhlon, O.; Vaknin, H.; Mishra, K.; D’Souza, J.; Marisat, M.; Sprecher, U.; Wald-Altman, S.; Dukhovny, A.; Raviv, Y.; Da’Adoosh, B.; et al. Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism. EMBO Mol. Med. 2021, 13, e14554. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1–e29. [Google Scholar] [CrossRef]
- Sarajlija, A.; Djordjevic, M.; Kecman, B.; Skakic, A.; Pavlovic, S.; Pasic, S.; Stojiljkovic, M. Impact of genotype on neutropenia in a large cohort of Serbian patients with glycogen storage disease type Ib. Eur. J. Med. Genet. 2019, 63, 103767. [Google Scholar] [CrossRef]
- Skakic, A.; Djordjevic, M.; Sarajlija, A.; Klaassen, K.; Tosic, N.; Kecman, B.; Ugrin, M.; Spasovski, V.; Pavlovic, S.; Stojiljkovic, M. Genetic characterization of GSD I in Serbian population revealed unexpectedly high incidence of GSD Ib and 3 novel SLC37A4 variants. Clin. Genet. 2017, 93, 350–355. [Google Scholar] [CrossRef]
- Melis, D.; Fulceri, R.; Parenti, G.; Marcolongo, P.; Gatti, R.; Parini, R.; Riva, E.; Della Casa, R.; Zammarchi, E.; Andria, G.; et al. Genotype/phenotype correlation in glycogen storage disease type 1b: A multicentre study and review of the literature. Eur. J. Pediatr. 2005, 164, 501–508. [Google Scholar] [CrossRef]
- Saban, O.S.; Pode-Shakked, B.; Abu-Libdeh, B.; Granot, M.; Barkai, G.; Haberman, Y.; Roterman, I.; Lahad, A.; Shouval, D.S.; Weiss, B.; et al. Glycogen Storage Disease type IA refractory to cornstarch: Can next generation sequencing offer a solution? Eur. J. Med. Genet. 2022, 65, 104518. [Google Scholar] [CrossRef]
- Scriver, C.R.; Waters, P.J. Monogenic traits are not simple: Lessons from phenylketonuria. Trends Genet. 1999, 15, 267–272. [Google Scholar] [CrossRef]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.; Scerif, G.; Wilde, L.; Beggs, A.; Stockton, J.; Sandhu, P.; Shelley, L.; Oliver, C.; McCleery, J. Genetic modifiers in rare disorders: The case of fragile X syndrome. Eur. J. Hum. Genet. 2020, 29, 173–183. [Google Scholar] [CrossRef]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; van der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 2010, 42, 801–805. [Google Scholar] [CrossRef]
- Radmilovic, M.; Zukic, B.; Petrovic, M.S.; Bartsakoulia, M.; Stankovic, B.; Kotur, N.; Dokmanovic, L.; Georgitsi, M.; Patrinos, G.P.; Pavlovic, S. Functional analysis of a novel KLF1 gene promoter variation associated with hereditary persistence of fetal hemoglobin. Ann. Hematol. 2012, 92, 53–58. [Google Scholar] [CrossRef]
- Mathis, T.; Poms, M.; Köfeler, H.; Gautschi, M.; Plecko, B.; Baumgartner, M.R.; Hochuli, M. Untargeted plasma metabolomics identifies broad metabolic perturbations in glycogen storage disease type I. J. Inherit. Metab. Dis. 2021, 45, 235–247. [Google Scholar] [CrossRef]
- Farah, B.L.; Yen, P.M.; Koeberl, D.D. Links between autophagy and disorders of glycogen metabolism—Perspectives on pathogenesis and possible treatments. Mol. Genet. Metab. 2019, 129, 3–12. [Google Scholar] [CrossRef]
- Koutsifeli, P.; Varma, U.; Daniels, L.J.; Annandale, M.; Li, X.; Neale, J.P.; Hayes, S.; Weeks, K.L.; James, S.; Delbridge, L.M.; et al. Glycogen-autophagy: Molecular machinery and cellular mechanisms of glycophagy. J. Biol. Chem. 2022, 298, 102093. [Google Scholar] [CrossRef]
- Bordi, M.; De Cegli, R.; Testa, B.; Nixon, R.A.; Ballabio, A.; Cecconi, F. A gene toolbox for monitoring autophagy transcription. Cell Death Dis. 2021, 12, 1044. [Google Scholar] [CrossRef]
- Zhao, H.; Tang, M.; Liu, M.; Chen, L. Glycophagy: An emerging target in pathology. Clin. Chim. Acta 2018, 484, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif-crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Kondomerkos, D.; Kalamidas, S.; Kotoulas, O. An electron microscopic and biochemical study of the effects of glucagon on glycogen autophagy in the liver and heart of newborn rats. Microsc. Res. Tech. 2004, 63, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kondomerkos, D.J.; Kalamidas, S.A.; Kotoulas, O.B.; Hann, A.C. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol. Histopathol. 2005, 20, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Kotoulas, A.O.; Kotoulas, O.B.; Kalamidas, S. An electron microscopic and biochemical study of the effects of cyclic 3′, 5′-AMP, ergotamine or propranolol on the lysosomes of newborn rat hepatocytes. Histol. Histopathol. 1991, 6, 421–426. [Google Scholar] [PubMed]
- An electron microscopic and biochemical study of the effects of propanolol on the glycogen autophagy in newborn rat hepatocytes. Histol. Histopathol. 2003, 18, 811–818. [CrossRef]
- Kalamidas, S.A.; Kondomerkos, D.J. The administration of nonmetabolizable glucose analogues fails to suppress the development of glycogen autophagy in newborn rat hepatocytes. Microsc. Res. Tech. 2010, 73, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Wray, H.; Gray, R. Cyclic AMP stimulation of membrane phosphorylation and Ca2+-activated, Mg2+-dependent ATPase in cardiac sarcoplasmic reticulum. Biochim. Biophys. Acta 1977, 461, 441–459. [Google Scholar] [CrossRef]
- Akin, B.L.; Hurley, T.D.; Chen, Z.; Jones, L.R. The Structural Basis for Phospholamban Inhibition of the Calcium Pump in Sarcoplasmic Reticulum. J. Biol. Chem. 2013, 288, 30181–30191. [Google Scholar] [CrossRef]
- Kotoulas, O.B.; Kalamidas, S.A.; Kondomerkos, D.J. Glycogen autophagy. Microsc. Res. Tech. 2004, 64, 10–20. [Google Scholar] [CrossRef]
- Kalamidas, S.; Kotoulas, O.; Hann, A. Studies on glycogen autophagy: Effects of phorbol myristate acetate, ionophore A23187, or phentolamine. Microsc. Res. Tech. 2002, 57, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; De Meyer, G.; Andries, L.; Herman, A.G.; Kockx, M.M. In Situ Detection of Starvation-induced Autophagy. J. Histochem. Cytochem. 2006, 54, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miao, J.-M. Ginkgolide K promotes astrocyte proliferation and migration after oxygen-glucose deprivation via inducing protective autophagy through the AMPK/mTOR/ULK1 signaling pathway. Eur. J. Pharmacol. 2018, 832, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, N.; Seto, S.W.; Chang, D.; Liang, H. Hydroxysafflor yellow a protects brain microvascular endothelial cells against oxygen glucose deprivation/reoxygenation injury: Involvement of inhibiting autophagy via class I PI3K/Akt/mTOR signaling pathway. Brain Res. Bull. 2018, 140, 243–257. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Q.; Chen, J.; Deng, B.; Qian, L.; Le, Y. PP2A Mediated AMPK Inhibition Promotes HSP70 Expression in Heat Shock Response. PLoS ONE 2010, 5, e13096. [Google Scholar] [CrossRef]
- Vauzour, D.; Corsini, S.; Muller, M.; Spencer, J.P. Inhibition of PP2A by hesperetin may contribute to Akt and ERK1/2 activation status in cortical neurons. Arch. Biochem. Biophys. 2018, 650, 14–21. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Caron, A.; Mouchiroud, M.; Gautier, N.; Labbé, S.M.; Villot, R.; Turcotte, L.; Secco, B.; Lamoureux, G.; Shum, M.; Gélinas, Y.; et al. Loss of hepatic DEPTOR alters the metabolic transition to fasting. Mol. Metab. 2017, 6, 447–458. [Google Scholar] [CrossRef]
- Bali, D.S.; Chen, Y.T.G.J. Glycogen Storage Disease Type I.; University of Washington: Washington, DC, USA, 2021. [Google Scholar]
- Ahn, H.-H.; Oh, Y.; Lee, H.; Lee, W.; Chang, J.-W.; Pyo, H.-K.; Nah, D.H.; Jung, Y.-K. Identification of glucose-6-phosphate transporter as a key regulator functioning at the autophagy initiation step. FEBS Lett. 2015, 589, 2100–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Heller, B.; Tagliabracci, V.S.; Zhai, L.; Irimia, J.M.; DePaoli-Roach, A.A.; Wells, C.D.; Skurat, A.V.; Roach, P.J. Starch Binding Domain-containing Protein 1/Genethonin 1 Is a Novel Participant in Glycogen Metabolism. J. Biol. Chem. 2010, 285, 34960–34971. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wells, C.D.; Roach, P.J. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem. Biophys. Res. Commun. 2011, 413, 420–425. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.M.; Martin, J.A.; Oxman, E.; Kasza, I.; Senn, K.A.; Dvinge, H. Alternative Splicing and Cleavage of GLUT8. Mol. Cell. Biol. 2020, 41, e00480-20. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A.J.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2013, 24, 24–41. [Google Scholar] [CrossRef]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.-I.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Chang, C.; Young, L.N.; Hurley, J.H. The BARA necessities of PtdIns 3-kinase activation in autophagy. Autophagy 2019, 15, 1122–1123. [Google Scholar] [CrossRef]
- Jatana, N.; Ascher, D.B.; Pires, D.E.; Gokhale, R.S.; Thukral, L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy 2019, 16, 239–255. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Thompson, C.B. Pathways of Apoptosis in Lymphocyte Development, Homeostasis, and Disease. Cell 2002, 109, S97–S107. [Google Scholar] [CrossRef]
- Sedger, L.M.; Katewa, A.; Pettersen, A.K.; Osvath, S.R.; Farrell, G.C.; Stewart, G.J.; Bendall, L.J.; Alexander, S.I. Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL and TRAIL double-deficient mice. Blood 2010, 115, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Lamhamedi-Cherradi, S.-E.; Zheng, S.-J.; Maguschak, K.A.; Peschon, J.; Chen, Y.H. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol. 2003, 4, 255–260. [Google Scholar] [CrossRef]
- Hongmei, Z. Extrinsic and Intrinsic Apoptosis Signal Pathway Review. In Apoptosis and medicine; InTechOpen: London, UK, 2012. [Google Scholar]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Honghong, Y.; Bing, H.; Yuan, Z.; Ling, S.R.H. Non-coding RNAs and Autophagy. In Autophagy: Biology and Diseases; Springer: Singapore, 2019; pp. 199–220. [Google Scholar]
- Ge, Y.-Y.; Shi, Q.; Zheng, Z.-Y.; Gong, J.; Zeng, C.; Yang, J.; Zhuang, S.-M. MicroRNA-100 promotes the autophagy of hepatocellular carcinoma cells by inhibiting the expression of mTOR and IGF-1R. Oncotarget 2014, 5, 6218–6228. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Song, J. Inhibition of autophagy potentiates the proliferation inhibition activity of microRNA-7 in human hepatocellular carcinoma cells. Oncol. Lett. 2017, 14, 3566–3572. [Google Scholar] [CrossRef]
- Chen, Y.; Liersch, R.; Detmar, M. The miR-290-295 cluster suppresses autophagic cell death of melanoma cells. Sci. Rep. 2012, 2, srep00808. [Google Scholar] [CrossRef]
- Li, W.; Yang, Y.; Ba, Z.; Li, S.; Chen, H.; Hou, X.; Ma, L.; He, P.; Jiang, L.; Li, L.; et al. MicroRNA-93 Regulates Hypoxia-Induced Autophagy by Targeting ULK1. Oxidative Med. Cell. Longev. 2017, 2017, 2709053. [Google Scholar] [CrossRef]
- Gu, D.-N.; Jiang, M.-J.; Mei, Z.; Dai, J.-J.; Dai, C.-Y.; Fang, C.; Huang, Q.; Tian, L. microRNA-7 impairs autophagy-derived pools of glucose to suppress pancreatic cancer progression. Cancer Lett. 2017, 400, 69–78. [Google Scholar] [CrossRef]
- Kim, D.; Hwang, H.-Y.; Kwon, H.J. Targeting Autophagy In Disease: Recent Advances In Drug Discovery. Expert Opin. Drug Discov. 2020, 15, 1045–1063. [Google Scholar] [CrossRef]
- Baek, S.H.; Kim, K.I. Epigenetic Control of Autophagy: Nuclear Events Gain More Attention. Mol. Cell 2017, 65, 781–785. [Google Scholar] [CrossRef]
- Möller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, N.; Brady, O.A.; Diab, H.I.; Martina, J.; Sun, L.; Huynh, T.; Lim, J.-A.; Zare, H.; Raben, N.; Ballabio, A.; et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016, 12, 1240–1258. [Google Scholar] [CrossRef]
- Klaassen, K.; Djordjevic, M.; Skakic, A.; Kecman, B.; Drmanac, R.; Pavlovic, S.; Stojiljkovic, M. Untreated PKU patients without intellectual disability: SHANK gene family as a candidate modifier. Mol. Genet. Metab. Rep. 2021, 29, 100822. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nho, K.; Kim, S.; Risacher, S.L.; Shen, L.; Bs, J.J.C.; Swaminathan, S.; Lin, H.; Ramanan, V.K.; Liu, Y.; Foroud, T.M.; et al. Protective variant for hippocampal atrophy identified by whole exome sequencing. Ann. Neurol. 2015, 77, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Mihaescu, R.; Pencina, M.J.; Alonso, A.; Lunetta, K.L.; Heckbert, S.R.; Benjamin, E.J.; Janssens, A.C.J. Incremental value of rare genetic variants for the prediction of multifactorial diseases. Genome Med. 2013, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.-Y.; Lu, F.-H.; Huang, X.-R.; Zhang, L.; Mao, W.; Yu, X.-Q.; Liu, X.-S.; Lan, H.-Y. Non-Coding RNAs as Biomarkers and Therapeutic Targets for Diabetic Kidney Disease. Front. Pharmacol. 2021, 11, 583528. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Goel, A. Non-coding RNAs as liquid biopsy biomarkers in cancer. Br. J. Cancer 2022, 126, 351–360. [Google Scholar] [CrossRef]
- Amanda, G.G. The Diversity of The Classification of Non-coding RNAs. J. Genom. Gene Study 2019, 2, 1. [Google Scholar]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA-Cancer Connection: The Beginning of a New Tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef]
- Akkoc, Y.; Gozuacik, D. MicroRNAs as major regulators of the autophagy pathway. Biochim. Biophys. Acta 2020, 1867, 118662. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2017, 46, D308–D314. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Shen, Q.; Feng, L.; Jin, H. Long non-coding RNAs involved in autophagy regulation. Cell Death Dis. 2017, 8, e3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, X.; Li, H.; Liu, J. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol. BioSyst. 2016, 12, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Moosavi, M.A.; Taheri, M. Exploring the role of non-coding RNAs in autophagy. Autophagy 2021, 18, 949–970. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhai, D.-S.; Huang, Q.; Chen, H.-L.; Zhang, Z. Tan, Q.-F. LncRNA DCST1-AS1 accelerates the proliferation, metastasis and autophagy of hepatocellular carcinoma cell by AKT/mTOR signaling pathways. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6091–6104. [Google Scholar] [CrossRef]
- Chen, C.-L.; Tseng, Y.-W.; Wu, J.-C.; Chen, G.-Y.; Lin, K.-C.; Hwang, S.-M.; Hu, Y.-C. Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials 2015, 44, 71–81. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Kuo, H.-C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef]
- Shang, J.; Chen, W.-M.; Liu, S.; Wang, Z.-H.; Wei, T.-N.; Chen, Z.-Z.; Wu, W.-B. CircPAN3 contributes to drug resistance in acute myeloid leukemia through regulation of autophagy. Leuk. Res. 2019, 85, 106198. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Yao, J.; Kang, J. Retracted: Circular RNA ACR relieves high glucose-aroused RSC96 cell apoptosis and autophagy via declining microRNA-145-3p. J. Cell. Biochem. 2019, 122, 1252. [Google Scholar] [CrossRef]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.-X. Autophagy in liver diseases: A review. Mol. Asp. Med. 2021, 82, 100973. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Pizarro, A.; Desviat, L.R. RNA solutions to treat inborn errors of metabolism. Mol. Genet. Metab. 2022, 136, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Pursell, N.; Gierut, J.; Zhou, W.; Dills, M.; Diwanji, R.; Gjorgjieva, M.; Saxena, U.; Yang, J.-S.; Shah, A.; Venkat, N.; et al. Inhibition of Glycogen Synthase II with RNAi Prevents Liver Injury in Mouse Models of Glycogen Storage Diseases. Mol. Ther. 2018, 26, 1771–1782. [Google Scholar] [CrossRef] [Green Version]

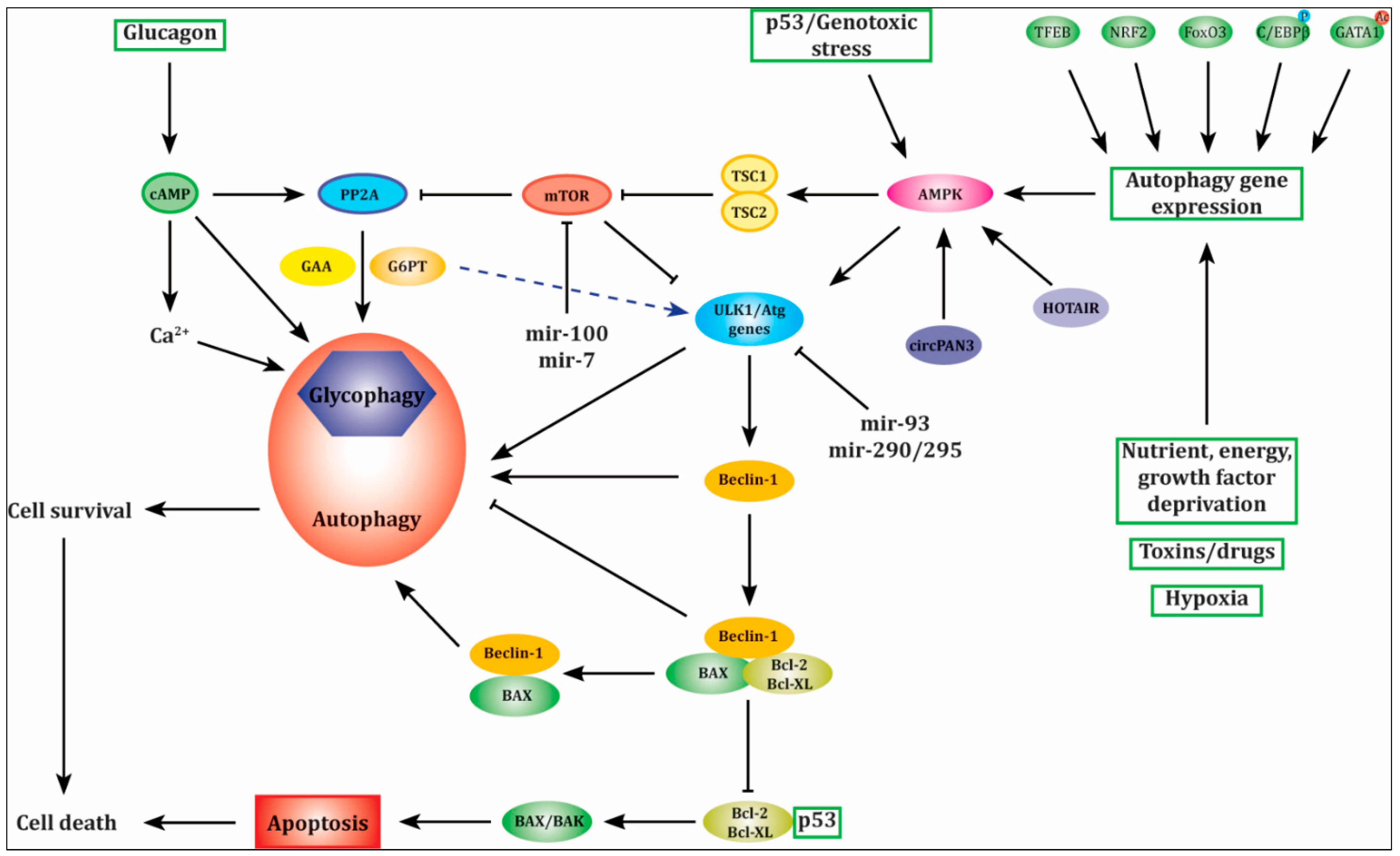

{kind=link}
{kind=link}
| Master Genes in the Transcriptional Regulation of Autophagy | TFEB family, NRF2, FoxO3 family, C/EBPβ family, GATA1 |
| miRNAs Involved in Autophagy Regulation | ULK1/2—miR-20a, miR-20b, miR-93, miR-10a, miR-106b, miR-17-5p, miR-290/295 mTOR—miR-17-5p, miR-30A/B/C, miR-129, miR-144, miR-409-3p, miR-100 RB1CC1—miR-20A/B, miR-224-3p Beclin-1—miR-17-5p, miR-124-3p, miR-216b, miR-376b Ambra1—miR-7, miR-23a UVRAG—miR-183, miR-216b, miR-351, miR-374a, miR-630,miR-1185 ATG14—miR-152 ATG12—miR-23a, miR-23b, miR-30, miR-214, miR-505-3p ATG5—miR-9a-5p, miR—142-3p, miR-181a, miR-224-3p, miR-638 ATG7—miR-17, miR-137, miR-210, miR-520b RAB7—miR-138-5p LAMP2—miR-21, miR-207, miR-224, miR-352, miR-373-5p, miR-379, miR-487b-5p RAB27/LAMP3—miR-205 |
| Long Non-Coding RNAs Involved in Autophagy Regulation | lncRNA H9, lncRNA NBR2, AD5-AlncRNA, lncRNA PTENP1, lncRNA MEG3, lncRNA ROR, lncRNA loc146880, lncRNA AC023115.3, lncRNA HOTAIRM1, lncRNA AK156230, lncRNA TGFB2, lncRNA GAS5, lncRNA HNF1A, lncRNA APF, lncRNA MALAT1, lncRNA HOTAIR, lncRNA PCGEM1, lncRNA Chast |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andjelkovic, M.; Skakic, A.; Ugrin, M.; Spasovski, V.; Klaassen, K.; Pavlovic, S.; Stojiljkovic, M. Crosstalk between Glycogen-Selective Autophagy, Autophagy and Apoptosis as a Road towards Modifier Gene Discovery and New Therapeutic Strategies for Glycogen Storage Diseases. Life 2022, 12, 1396. https://doi.org/10.3390/life12091396
Andjelkovic M, Skakic A, Ugrin M, Spasovski V, Klaassen K, Pavlovic S, Stojiljkovic M. Crosstalk between Glycogen-Selective Autophagy, Autophagy and Apoptosis as a Road towards Modifier Gene Discovery and New Therapeutic Strategies for Glycogen Storage Diseases. Life. 2022; 12(9):1396. https://doi.org/10.3390/life12091396
Chicago/Turabian StyleAndjelkovic, Marina, Anita Skakic, Milena Ugrin, Vesna Spasovski, Kristel Klaassen, Sonja Pavlovic, and Maja Stojiljkovic. 2022. "Crosstalk between Glycogen-Selective Autophagy, Autophagy and Apoptosis as a Road towards Modifier Gene Discovery and New Therapeutic Strategies for Glycogen Storage Diseases" Life 12, no. 9: 1396. https://doi.org/10.3390/life12091396
APA StyleAndjelkovic, M., Skakic, A., Ugrin, M., Spasovski, V., Klaassen, K., Pavlovic, S., & Stojiljkovic, M. (2022). Crosstalk between Glycogen-Selective Autophagy, Autophagy and Apoptosis as a Road towards Modifier Gene Discovery and New Therapeutic Strategies for Glycogen Storage Diseases. Life, 12(9), 1396. https://doi.org/10.3390/life12091396