
P16INK4A—More Than a Senescence Marker




Abstract
1. Introduction
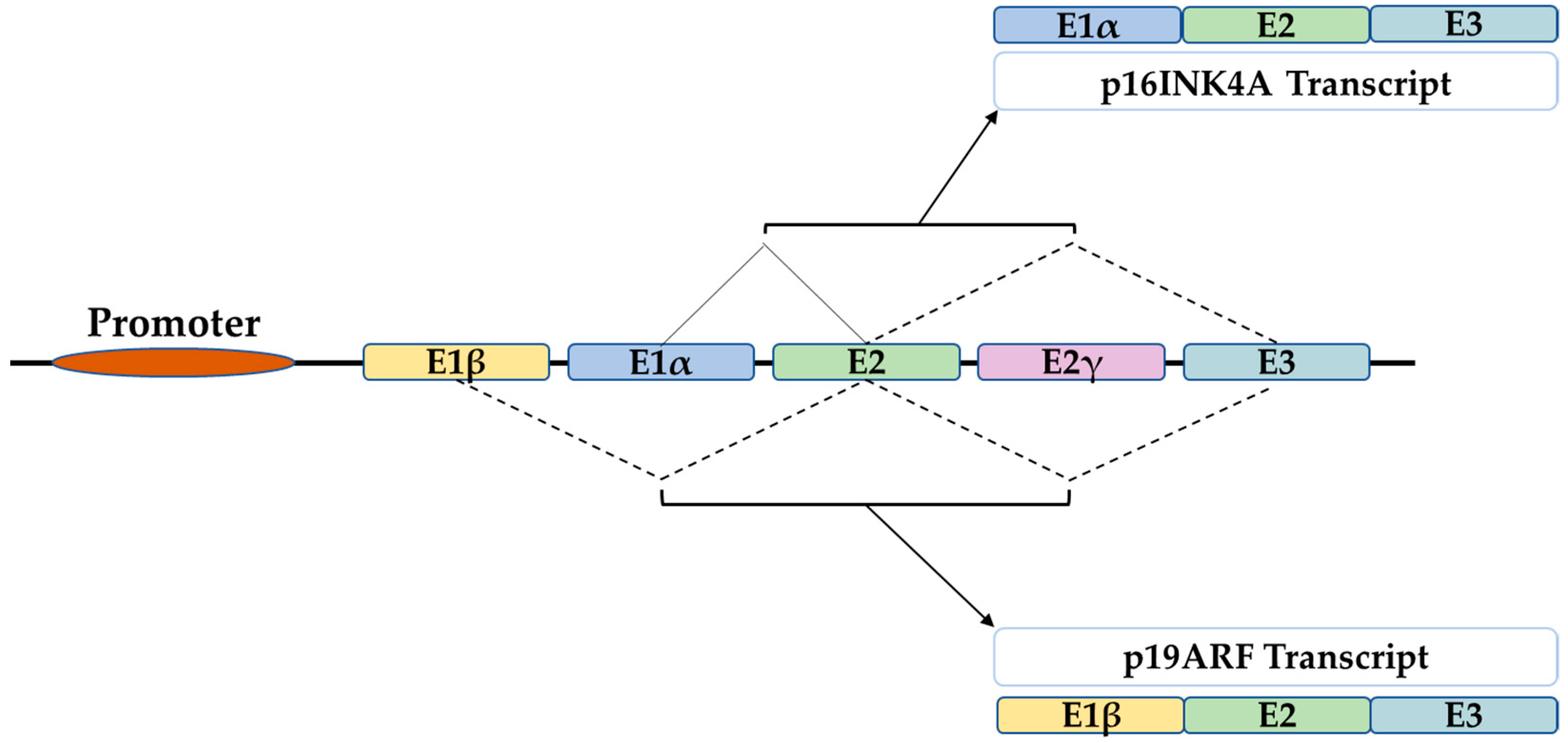
2. The P16 Gene
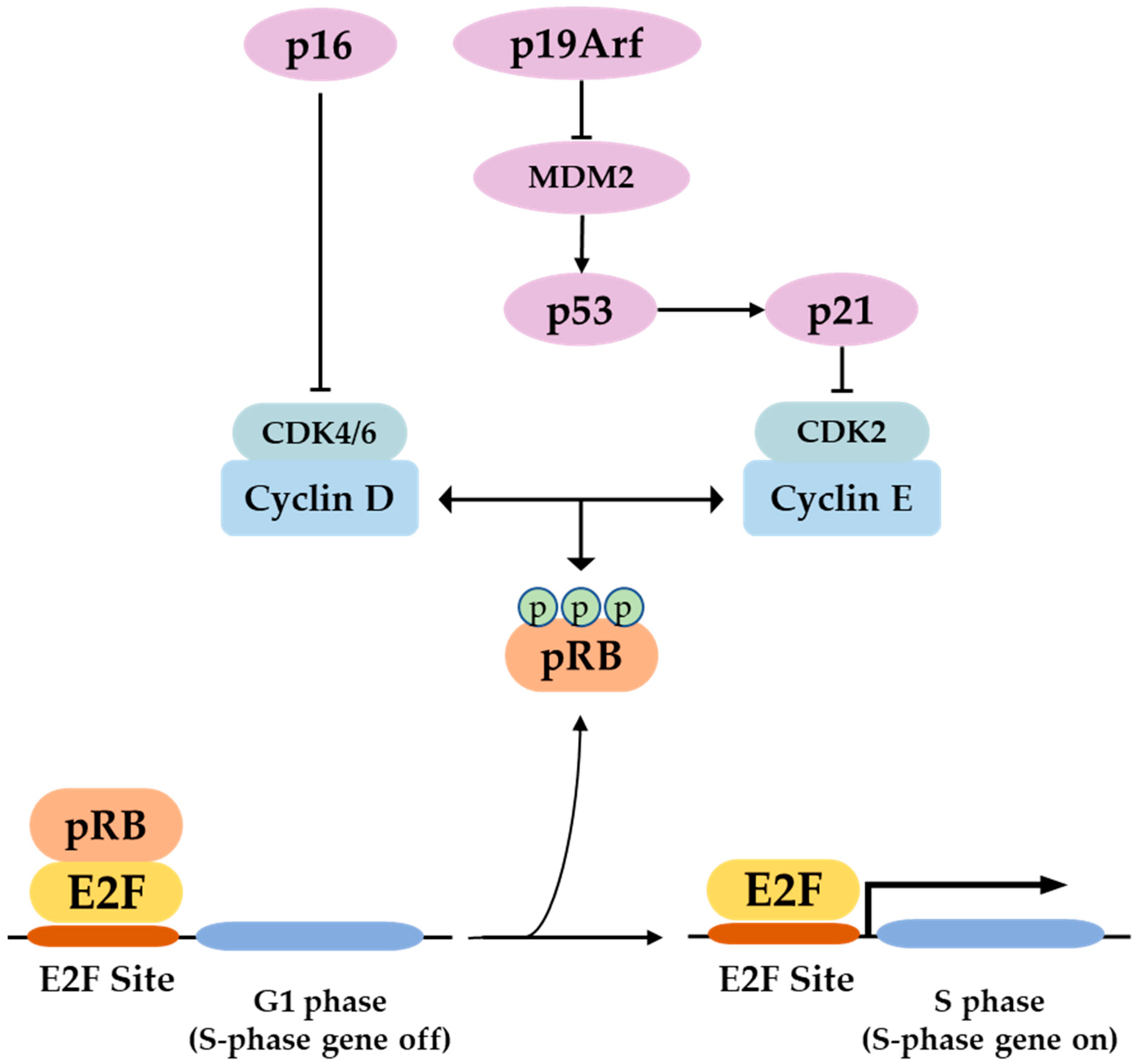
3. P16 Function and Regulation of Expression
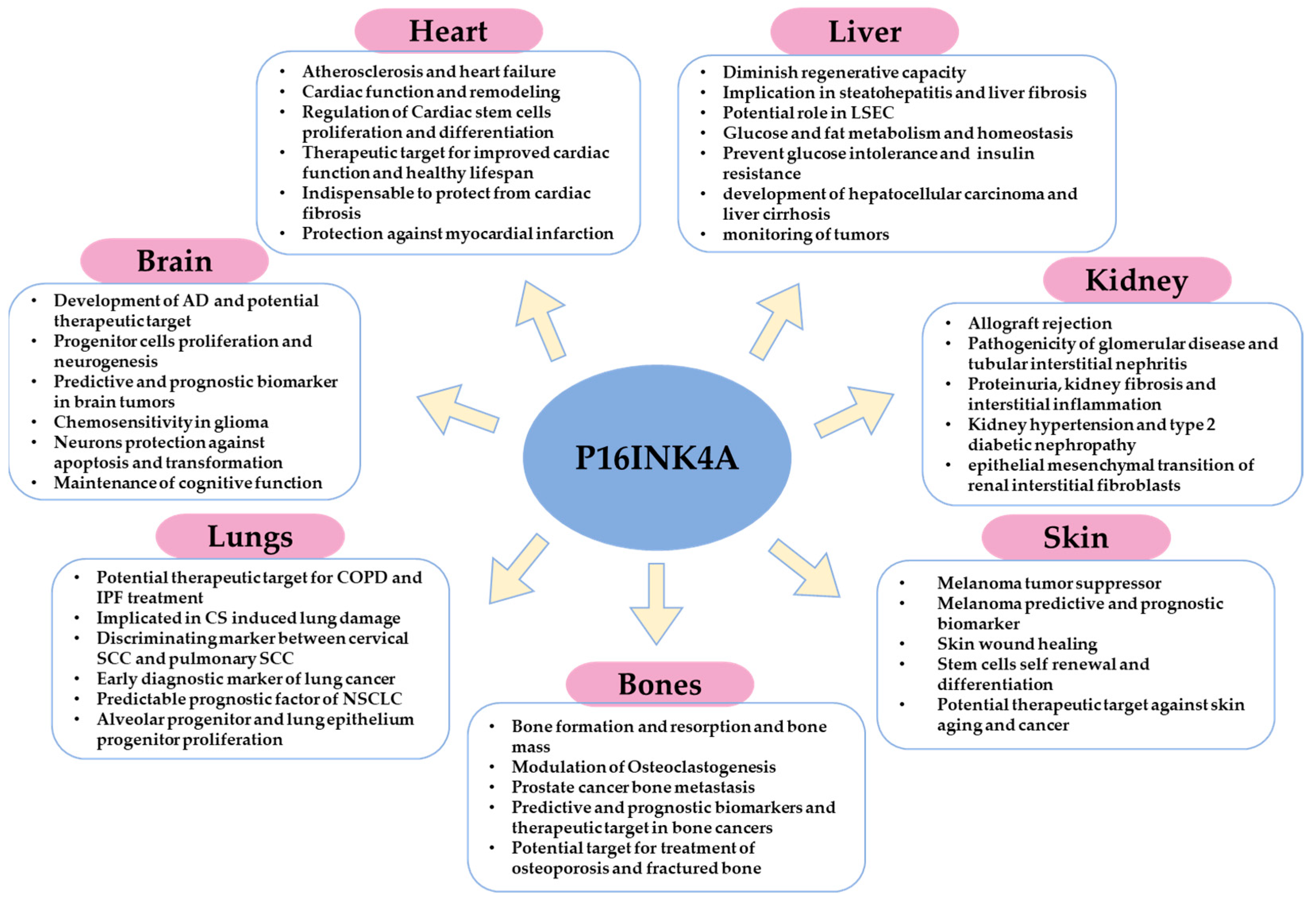
4. Role of p16 in Different Tissues and Organs: Cancer, Physiology, and Pathophysiology
4.1. In the Skin
4.2. In the Bones
4.3. In the Lungs
4.4. In the Brain
4.5. In the Heart
4.6. In the Kidney
4.7. In the Liver

{kind=link}
{kind=link}
{kind=link}
| Process | Model and/or Tissue | Potential Role/Function | References |
|---|---|---|---|
| Intervertebral disc damage | Mouse and human intervertebral disc tissues | P16 is a potential therapeutic target for intervertebral disc damage relief. | [48] |
| Wound healing | P16-3MR model (fibroblasts, endothelial cells, and keratinocytes) | Accelerate wound closure and re-epithelialization | [43] |
| Tumorigenesis and tumor suppression | Skin, bone, lung, liver, and brain cancer patients tissues and immunohistopathology | Implicated in tumor development, progression, and metastasis Predictive and prognostic marker Therapeutic target Increases chemosensitivity | [39,40,41,42,72,105,106,107,108,111,112,134,135,136,150,151,152,153,168,169,170,205,206] |
| Stem cell self-renewal and differentiation | Skin, lung, bone, brain, and heart stem cells | Balanced expression of p16 is a prerequisite for stem cells proliferation and differentiation. Therapeutic approach for maintenance of regenerative capacity | [46,92,101,115,116,117,118,119,138,149,155,156,179] |
| Cellular senescence | Primary mouse fibroblasts and melanocytes | Target for oncogene-induced senescence bypass and aging | [37,38] |
| Bone homeostasis | P16-3MR and p16-INK-ATTAC mouse model | Maintenance of bone mass Orchestration of osteoblast and osteoclast function | [123,132,133] |
| Bone fracture healing | Geriatric Mouse model (p16-/- and WT) | P16-deletion stimulated osteoblastogenesis and vascularization and accelerated bone fracture healing | [138] |
| Muscle injury | Acute muscle injury (AIM) mouse model | Tissue regeneration | [45] |
| Osteoporosis | Ovariectomized p16-/- and WT mice | Potential therapeutic target to prevent estrogen-induced osteoporosis | [124,125,126,127,137] |
| COPD | Lung alveolar and lung epithelial cells in mice and human | Implicated in COPD severity Potential therapeutic target | [140,141,142,149] |
| Cervical SCC and pulmonary SCC | Human cancer patients | Discriminating biomarker | [150] |
| Oxidative stress | Fibroblasts, keratinocytes, and melanocytes | P16 regulates oxidative stress and ROS production as pRB-independent tumor suppression mechanism | [72,204] |
| Mitochondrial biogenesis | Primary mouse fibroblasts, human melanocytes, A375 melanoma cells | P16 balances mitochondrial structure and function | [111] |
| Alzheimer’s disease (AD) | Alzheimer’s disease patients and mouse model | Implicated in AD severity and development Therapeutic target | [159,160,161,166] |
| Lung injury | P16-/- and WT mouse model (lung epithelium) | P16 protects against lungs injury | [154] |
| Cardiac fibrosis | p16-CreERT2-tdTomato mouse model | P16-positive cells removal induces cardiac fibrosis | [83] |
| Myocardial Infarction | Mice | Indispensable for maintenance of cardiac function and cardiac remodeling after infarction | [181] |
| Glucose metabolism and homeostasis | Super-INK4A/ARF mice model | Prevented the development of glucose intolerance with aging Protective role against age-induced insulin resistance | [198] |
| Liver fibrosis | INK-ATTAC mouse model | Therapeutic approach for treatment of liver fibrosis | [83,88,204] |
| Fat metabolism | Mouse model and primary hepatocytes | Regulate fasting-induced fatty acid oxidation and lipid droplet accumulation in the liver | [201] |
| Development | Young mice brain, heart, kidney, and liver | Dynamic p16 expression detected in embryonic mice organs, reflecting a potential role in embryonic development | [120] |
5. The Role of p16 in Development
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Stone, S.; Jiang, P.; Dayananth, P.; Tavtigian, S.V.; Katcher, H.; Parry, D.; Peters, G.; Kamb, A. Complex structure and regulation of the P16 (MTS1) locus. Cancer Res. 1995, 55, 2988–2994. [Google Scholar]
- Mao, L.; Merlo, A.; Bedi, G.; Shapiro, G.I.; Edwards, C.D.; Rollins, B.J.; Sidransky, D. A novel p16INK4A transcript. Cancer Res. 1995, 55, 2995–2997. [Google Scholar] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- López-Domínguez, J.A.; Rodríguez-López, S.; Ahumada-Castro, U.; Desprez, P.-Y.; Konovalenko, M.; Laberge, R.-M.; Cárdenas, C.; Villalba, J.M.; Campisi, J. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging 2021, 13, 13380–13392. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Ohtani, N.; Mann, D.J.; Hara, E. Cellular senescence: Its role in tumor suppression and aging. Cancer Sci. 2009, 100, 792–797. [Google Scholar] [CrossRef]
- Understanding the Dynamics of the Aging Process National Institute on Aging. Available online: http://www.nia.nih.gov/about/aging-strategic-directions-research/understanding-dynamics-aging (accessed on 28 July 2022).
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Ramly, E.; Kaafarani, H.M.A.; Velmahos, G.C. The effect of aging on pulmonary function: Implications for monitoring and support of the surgical and trauma patient. Surg. Clin. North Am. 2015, 95, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.R. Renal function in aging. J. Am. Geriatr. Soc. 1989, 37, 791–800. [Google Scholar] [CrossRef]
- Tung, S.; Iqbal, J. Evolution, Aging, and Osteoporosis. Ann. N. Y. Acad. Sci. 2007, 1116, 499–506. [Google Scholar] [CrossRef]
- Misra, D.; Seo, P.H.; Cohen, H.J. Aging and cancer. Clin. Adv. Hematol. Oncol. 2004, 2, 457–465. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef]
- Best, B.P. Nuclear DNA Damage as a Direct Cause of Aging. Rejuvenation Res. 2009, 12, 199–208. [Google Scholar] [CrossRef]
- Freitas, A.A.; de Magalhães, J.P. A review and appraisal of the DNA damage theory of ageing. Mutat. Res./Rev. Mutat. Res. 2011, 728, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Collins, C.J.; Sedivy, J.M. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell 2003, 2, 145–150. [Google Scholar] [CrossRef]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic Cooperation between p16INK4a and p21Waf1/Cip1 in the Onset of Cellular Senescence and Tumor Suppression In vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Bates, S.; Mann, D.J.; Peters, G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. EMBO J. 1995, 14, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Dev. Camb. Engl. 2019, 146, dev151837. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Buj, R.; Leon, K.E.; Anguelov, M.A.; Aird, K.M. Suppression of p16 alleviates the senescence-associated secretory phenotype. Aging 2021, 13, 3290–3312. [Google Scholar] [CrossRef]
- Buj, R.; Chen, C.-W.; Dahl, E.S.; Leon, K.E.; Kuskovsky, R.; Maglakelidze, N.; Navaratnarajah, M.; Zhang, G.; Doan, M.T.; Jiang, H.; et al. Suppression of p16 Induces mTORC1-Mediated Nucleotide Metabolic Reprogramming. Cell Rep. 2019, 28, 1971–1980e8. [Google Scholar] [CrossRef]
- Damsky, W.; Micevic, G.; Meeth, K.; Muthusamy, V.; Curley, D.P.; Santhanakrishnan, M.; Erdelyi, I.; Platt, J.T.; Huang, L.; Theodosakis, N.; et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 2015, 27, 41–56. [Google Scholar] [CrossRef]
- Dankort, D.; Filenova, E.; Collado, M.; Serrano, M.; Jones, K.; McMahon, M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007, 21, 379–384. [Google Scholar] [CrossRef]
- Goel, V.K.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.S.; Hinds, P.W.; Haluska, F.G. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009, 28, 2289–2298. [Google Scholar] [CrossRef]
- Haferkamp, S.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell 2008, 7, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, E.; Omobono, J.D.; Jones, J.C.; Rheinwald, J.G. Co-expression of p16INK4A and laminin 5 by keratinocytes: A wound-healing response coupling hypermotility with growth arrest that goes awry during epithelial neoplastic progression. J. Investig. Dermatol. Symp. Proc. 2005, 10, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Chikenji, T.S.; Saito, Y.; Konari, N.; Nakano, M.; Mizue, Y.; Otani, M.; Fujimiya, M. p16INK4A-expressing mesenchymal stromal cells restore the senescence–clearance–regeneration sequence that is impaired in chronic muscle inflammation. eBioMedicine 2019, 44, 86–97. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Che, H.; Li, J.; Li, Y.; Ma, C.; Liu, H.; Qin, J.; Dong, J.; Zhang, Z.; Xian, C.J.; Miao, D.; et al. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. eLife 2020, 9, e52570. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Huang, X.; Tang, J.; Zhang, S.; Li, Y.; Liu, X.; He, L.; Ju, Z.; Lui, K.O.; et al. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. 2018, 28, 775–778. [Google Scholar] [CrossRef]
- Hosako, H.; Francisco, L.E.; Martin, G.S.; Mirkes, P.E. The roles of p53 and p21 in normal development and hyperthermia-induced malformations. Birth Defects Res. B. Dev. Reprod. Toxicol. 2009, 86, 40–47. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Soares, H.; Herzog, K.H.; Morgan, J.; Sherr, C.J.; Roussel, M.F. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997, 8, 1139–1150. [Google Scholar] [PubMed]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M. The tumor suppressor protein p16INK4a. Exp. Cell Res. 1997, 237, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Komata, T.; Kanzawa, T.; Takeuchi, H.; Germano, I.M.; Schreiber, M.; Kondo, Y.; Kondo, S. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br. J. Cancer 2003, 88, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M.-D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Cilluffo, D.; Barra, V.; Di Leonardo, A. P14ARF: The Absence that Makes the Difference. Genes 2020, 11, 824. [Google Scholar] [CrossRef]
- Liggett, W.H.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Cat and Mouse Games That Genes, Viruses, and Cells Play. Cell 1997, 88, 573–575. [Google Scholar] [CrossRef]
- Ahlander, J.; Bosco, G. The RB/E2F pathway and regulation of RNA processing. Biochem. Biophys. Res. Commun. 2009, 384, 280–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Nallar, S.C.; Raha, A.; Kalakonda, S.; Velalar, C.N.; Reddy, S.P.; Kalvakolanu, D.V. GRIM-19 and p16(INK4a) synergistically regulate cell cycle progression and E2F1-responsive gene expression. J. Biol. Chem. 2010, 285, 27545–27552. [Google Scholar] [CrossRef]
- Li, Y.; Nichols, M.A.; Shay, J.W.; Xiong, Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 1994, 54, 6078–6082. [Google Scholar]
- Sanchez-Cespedes, M.; Reed, A.L.; Buta, M.; Wu, L.; Westra, W.H.; Herman, J.G.; Yang, S.C.; Jen, J.; Sidransky, D. Inactivation of the INK4A/ARF locus frequently coexists with TP53 mutations in non-small cell lung cancer. Oncogene 1999, 18, 5843–5849. [Google Scholar] [CrossRef]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.-C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16INK4A tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wong, P.K.Y. Oxidative stress is linked to ERK1/2-p16 signaling-mediated growth defect in ATM-deficient astrocytes. J. Biol. Chem. 2009, 284, 14396–14404. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, E.; Turner, S.L.; Harju, S.; Miyazaki, S.; Kashiwagi, M.; Koh, J.; Serizawa, H. Regulation of CDK7-carboxyl-terminal domain kinase activity by the tumor suppressor p16(INK4A) contributes to cell cycle regulation. Mol. Cell. Biol. 2000, 20, 7726–7734. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Choi, H.S.; Ko, K.; Cho, Y.-Y.; Zhu, F.; Kang, B.S.; Ermakova, S.P.; Ma, W.-Y.; Bode, A.M.; Dong, Z. The tumor suppressor p16(INK4a) prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nat. Struct. Mol. Biol. 2005, 12, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, H. Cyclin-dependent kinase inhibitor p16INK4A inhibits phosphorylation of RNA polymerase II by general transcription factor TFIIH. J. Biol. Chem. 1998, 273, 5427–5430. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef]
- Graves, B.J.; Petersen, J.M. Specificity within the ets family of transcription factors. Adv. Cancer Res. 1998, 75, 1–57. [Google Scholar] [CrossRef]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.G.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef]
- Xiu, M.; Kim, J.; Sampson, E.; Huang, C.-Y.; Davis, R.J.; Paulson, K.E.; Yee, A.S. The Transcriptional Repressor HBP1 Is a Target of the p38 Mitogen-Activated Protein Kinase Pathway in Cell Cycle Regulation. Mol. Cell. Biol. 2003, 23, 8890–8901. [Google Scholar] [CrossRef]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99e6. [Google Scholar] [CrossRef] [PubMed]
- Matheu, A.; Pantoja, C.; Efeyan, A.; Criado, L.M.; Martín-Caballero, J.; Flores, J.M.; Klatt, P.; Serrano, M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004, 18, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar] [CrossRef]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Doolittle, M.; Tchkonia, T.; Kirkland, J.L.; LeBrasseur, N.K.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022, 21, e13602. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.-J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077e8. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Le Goff, O.; Soysouvanh, F.; Vasseur, F.; Tanou, M.; Nguyen, C.; Amrouche, L.; Le Guen, J.; Saltel-Fulero, O.; Meunier, T.; et al. Glomerular endothelial cell senescence drives age-related kidney disease through PAI-1. EMBO Mol. Med. 2021, 13, e14146. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.; Dong, Q.; Wang, D.; Chang, J.; Wiley, C.; Demaria, M.; Lee, J.; Kang, J.; Niedernhofer, L.J.; Robbins, P.D.; et al. Systemic clearance of p16INK4a -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 2019, 18, e12927. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Jeon, O.H.; Mehdipour, M.; Gil, T.-H.; Kang, M.; Aguirre, N.W.; Robinson, Z.R.; Kato, C.; Etienne, J.; Lee, H.G.; Alimirah, F.; et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat. Metab. 2022, 4, 995–1006. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal Stem Cells of the Skin. Annu. Rev. Cell Dev. Biol. 2006, 22, 339–373. [Google Scholar] [CrossRef]
- Kligman, A.M. Perspectives and Problems in Cutaneous Gerontology. J. Investig. Dermatol. 1979, 73, 39–46. [Google Scholar] [CrossRef]
- Montagna, W.; Carlisle, K. Structural Changes in Aging Human Skin. J. Investig. Dermatol. 1979, 73, 47–53. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Recent advances on skin-resident stem/progenitor cell functions in skin regeneration, aging and cancers and novel anti-aging and cancer therapies. J. Cell. Mol. Med. 2010, 14, 116–134. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-β1 signaling pathway. J. Cell Sci. 2005, 118, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-induced Senescence in Human Keratinocytes Requires a Functional Insulin-like Growth Factor-1 Receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- McCart, E.A.; Thangapazham, R.L.; Lombardini, E.D.; Mog, S.R.; Panganiban, R.A.M.; Dickson, K.M.; Mansur, R.A.; Nagy, V.; Kim, S.-Y.; Selwyn, R.; et al. Accelerated senescence in skin in a murine model of radiation-induced multi-organ injury. J. Radiat. Res. 2017, 58, 636–646. [Google Scholar] [CrossRef]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef]
- Scholes, A.G.; Liloglou, T.; Maloney, P.; Hagan, S.; Nunn, J.; Hiscott, P.; Damato, B.E.; Grierson, I.; Field, J.K. Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2472–2477. [Google Scholar]
- Castellano, M.; Pollock, P.M.; Walters, M.K.; Sparrow, L.E.; Down, L.M.; Gabrielli, B.G.; Parsons, P.G.; Hayward, N.K. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res. 1997, 57, 4868–4875. [Google Scholar]
- Funk, J.O.; Schiller, P.I.; Barrett, M.T.; Wong, D.J.; Kind, P.; Sander, C.A. p16INK4a expression is frequently decreased and associated with 9p21 loss of heterozygosity in sporadic melanoma. J. Cutan. Pathol. 1998, 25, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Straume, O.; Sviland, L.; Akslen, L.A. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1845–1853. [Google Scholar]
- Mihic-Probst, D.; Mnich, C.D.; Oberholzer, P.A.; Seifert, B.; Sasse, B.; Moch, H.; Dummer, R. p16 expression in primary malignant melanoma is associated with prognosis and lymph node status. Int. J. Cancer 2006, 118, 2262–2268. [Google Scholar] [CrossRef]
- Tyagi, E.; Liu, B.; Li, C.; Liu, T.; Rutter, J.; Grossman, D. Loss of p16INK4A stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget 2017, 8, 55848–55862. [Google Scholar] [CrossRef][Green Version]
- Al-Khalaf, H.H.; Mohideen, P.; Nallar, S.C.; Kalvakolanu, D.V.; Aboussekhra, A. The cyclin-dependent kinase inhibitor p16INK4a physically interacts with transcription factor Sp1 and cyclin-dependent kinase 4 to transactivate microRNA-141 and microRNA-146b-5p spontaneously and in response to ultraviolet light-induced DNA damage. J. Biol. Chem. 2013, 288, 35511–35525. [Google Scholar] [CrossRef]
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.; Conroy, S.; Russell, T.; Gabrielli, B. Ultraviolet Radiation Induces p16CDKN2A Expression in Human Skin1. Cancer Res. 1999, 59, 4185–4189. [Google Scholar] [PubMed]
- Adam, R.C.; Fuchs, E. The Yin and Yang of Chromatin Dynamics In Stem Cell Fate Selection. Trends Genet. 2016, 32, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic regulation of epidermal differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef]
- Safwan-Zaiter, H.; Wagner, N.; Michiels, J.-F.; Wagner, K.-D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells 2022, 11, 541. [Google Scholar] [CrossRef]
- Langlands, K.; Down, G.A.; Kealey, T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res. 2000, 60, 5929–5933. [Google Scholar]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-Osteoclast Interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; O’Brien, C.A. The Role of Osteocytes in Age-Related Bone Loss. Curr. Osteoporos. Rep. 2016, 14, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef]
- Glatt, V.; Canalis, E.; Stadmeyer, L.; Bouxsein, M.L. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J. Bone Miner. Res. 2007, 22, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, M.; Almeida, M.; Robling, A.G.; Kim, H.-N.; Xiong, J.; Thostenson, J.D.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2017, 2, 93771. [Google Scholar] [CrossRef]
- Ucer, S.; Iyer, S.; Kim, H.-N.; Han, L.; Rutlen, C.; Allison, K.; Thostenson, J.D.; de Cabo, R.; Jilka, R.L.; O’Brien, C.; et al. The Effects of Aging and Sex Steroid Deficiency on the Murine Skeleton Are Independent and Mechanistically Distinct. J. Bone Miner. Res. 2017, 32, 560–574. [Google Scholar] [CrossRef]
- Marie, P.J. Bone Cell Senescence: Mechanisms and Perspectives. J. Bone Miner. Res. 2014, 29, 1311–1321. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef]
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-N.; Chang, J.; Iyer, S.; Han, L.; Campisi, J.; Manolagas, S.C.; Zhou, D.; Almeida, M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 2019, 18, e12923. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Chen, X.; Huang, C.; Yang, C.; Situ, M.; Zhou, Q.; Ling, Y.; Huang, H.; Huang, M.; Zhang, Y.; et al. UBE2S as a novel ubiquitinated regulator of p16 and β-catenin to promote bone metastasis of prostate cancer. Int. J. Biol. Sci. 2022, 18, 3528–3543. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.S.; Thomas, R.G.; Passant, C.D. Do patients with p16-positive oropharyngeal squamous cell carcinoma get more bone metastasis than p16-negative patients? J. Laryngol. Otol. 2018, 132, 429–433. [Google Scholar] [CrossRef]
- Righi, A.; Gambarotti, M.; Sbaraglia, M.; Sisto, A.; Ferrari, S.; Dei Tos, A.P.; Picci, P. p16 expression as a prognostic and predictive marker in high-grade localized osteosarcoma of the extremities: An analysis of 357 cases. Hum. Pathol. 2016, 58, 15–23. [Google Scholar] [CrossRef]
- Li, J.; Karim, M.A.; Che, H.; Geng, Q.; Miao, D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am. J. Transl. Res. 2020, 12, 672–683. [Google Scholar]
- Ding, Q.; Liu, H.; Liu, L.; Ma, C.; Qin, H.; Wei, Y.; Ren, Y. Deletion of p16 accelerates fracture healing in geriatric mice. Am. J. Transl. Res. 2021, 13, 11107. [Google Scholar]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef]
- Fukuchi, Y. The aging lung and chronic obstructive pulmonary disease: Similarity and difference. Proc. Am. Thorac. Soc. 2009, 6, 570–572. [Google Scholar] [CrossRef]
- John-Schuster, G.; Günter, S.; Hager, K.; Conlon, T.M.; Eickelberg, O.; Yildirim, A.Ö. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget 2016, 7, 30068–30083. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Selman, M.; Buendía-Roldán, I.; Pardo, A. Aging and Pulmonary Fibrosis. Rev. Investig. Clin. 2016, 68, 75–83. [Google Scholar]
- Venosa, A. Senescence in Pulmonary Fibrosis: Between Aging and Exposure. Front. Med. 2020, 7, 606462. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, F.; You, R.; Hee Gu, B.; Corry, D.B. Cigarette Smoke and DNA Cleavage Promote Lung Inflammation and Emphysema. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 222–233. [Google Scholar]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.L.; Glaser, H.; Cagley, J.R.; Brown, C.O.; Matsumoto, E.; Aykin-Burns, N.; Spitz, D.R.; Oshima, J.; et al. Cigarette Smoke Induces Cellular Senescence via Werner’s Syndrome Protein Down-regulation. Am. J. Respir. Crit. Care Med. 2009, 179, 279–287. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 9023. [Google Scholar] [CrossRef]
- Cottage, C.T.; Peterson, N.; Kearley, J.; Berlin, A.; Xiong, X.; Huntley, A.; Zhao, W.; Brown, C.; Migneault, A.; Zerrouki, K.; et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun. Biol. 2019, 2, 307. [Google Scholar] [CrossRef]
- Wang, C.-W.; Wu, T.-I.; Yu, C.-T.; Wu, Y.-C.; Teng, Y.-H.; Chin, S.-Y.; Lai, C.-H.; Chen, T.-C. Usefulness of p16 for differentiating primary pulmonary squamous cell carcinoma from cervical squamous cell carcinoma metastatic to the lung. Am. J. Clin. Pathol. 2009, 131, 715–722. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11891–11896. [Google Scholar] [CrossRef]
- Okamoto, A.; Hussain, S.P.; Hagiwara, K.; Spillare, E.A.; Rusin, M.R.; Demetrick, D.J.; Serrano, M.; Hannon, G.J.; Shiseki, M.; Zariwala, M.; et al. Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 Genes in Primary and Metastatic Lung Cancer1. Cancer Res. 1995, 55, 1448–1451. [Google Scholar] [PubMed]
- Tong, J.; Sun, X.; Cheng, H.; Zhao, D.; Ma, J.; Zhen, Q.; Cao, Y.; Zhu, H.; Bai, J. Expression of p16 in non-small cell lung cancer and its prognostic significance: A meta-analysis of published literatures. Lung Cancer 2011, 74, 155–163. [Google Scholar] [CrossRef] [PubMed]
- p16 Regulation of Lung Epithelial Cell Growth, Repair after Injury and Transformation—ProQuest. Available online: https://www.proquest.com/openview/c4b38b5ea3ce9a758bbacff031039f5c/1?pq-origsite=gscholar&cbl=18750&diss=y (accessed on 28 July 2022).
- De Mochel, N.R.; Cheong, K.N.; Cassandras, M.; Wang, C.; Krasilnikov, M.; Matatia, P.; Molofsky, A.; Campisi, J.; Peng, T. Sentinel p16INK4a+ cells in the basement membrane form a reparative niche in the lung 2020. bioRxiv 2020. [CrossRef]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Lok, K.; Zhao, H.; Shen, H.; Wang, Z.; Gao, X.; Zhao, W.; Yin, M. Characterization of the APP/PS1 mouse model of Alzheimer’s disease in senescence accelerated background. Neurosci. Lett. 2013, 557, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Dorigatti, A.O.; Riordan, R.; Yu, Z.; Ross, G.; Wang, R.; Reynolds-Lallement, N.; Magnusson, K.; Galvan, V.; Perez, V.I. Brain cellular senescence in mouse models of Alzheimer’s disease. GeroScience 2022, 44, 1157–1168. [Google Scholar] [CrossRef]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-Dependent Neurofibrillary Tangle Formation, Neuron Loss, and Memory Impairment in a Mouse Model of Human Tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- Mv, S.; Hv, V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. Tripartite synapses: Roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 2009, 57, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; García-Matas, S.; Rodriguez-Farre, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Jen, J.; Harper, J.W.; Bigner, S.H.; Bigner, D.D.; Papadopoulos, N.; Markowitz, S.; Willson, J.K.V.; Kinzler, K.W.; Vogelstein, B. Deletion of p16 and p15 Genes in Brain Tumors1. Cancer Res. 1994, 54, 6353–6358. [Google Scholar]
- Ueki, K.; Ono, Y.; Henson, J.W.; Efird, J.T.; von Deimling, A.; Louis, D.N. CDKN2/p16 or RB Alterations Occur in the Majority of Glioblastomas and Are Inversely Correlated1. Cancer Res. 1996, 56, 150–153. [Google Scholar]
- Park, J.W.; Kang, J.; Lim, K.Y.; Kim, H.; Kim, S.-I.; Won, J.K.; Park, C.-K.; Park, S.-H. The prognostic significance of p16 expression pattern in diffuse gliomas. J. Pathol. Transl. Med. 2021, 55, 102–111. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Puduvalli, V.K.; Martin-Duque, P.; Perez-Soler, R.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. Adenovirus-mediated p16 transfer to glioma cells induces G1 arrest and protects from paclitaxel and topotecan: Implications for therapy. Int. J. Oncol. 1998, 12, 665–674. [Google Scholar] [CrossRef]
- Kranenburg, O.; van der Eb, A.J.; Zantema, A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 1996, 15, 46–54. [Google Scholar] [CrossRef]
- Kfoury, N.; Sun, T.; Yu, K.; Rockwell, N.; Tinkum, K.L.; Qi, Z.; Warrington, N.M.; McDonald, P.; Roy, A.; Weir, S.J.; et al. Cooperative p16 and p21 action protects female astrocytes from transformation. Acta Neuropathol. Commun. 2018, 6, 12. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, X.; Cao, J.; Zhang, W.; Lu, C.; Chen, X. Dihydromyricetin attenuates D-galactose-induced brain aging of mice via inhibiting oxidative stress and neuroinflammation. Neurosci. Lett. 2021, 756, 135963. [Google Scholar] [CrossRef]
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and Death of Primitive Cells and Myocytes Lead to Premature Cardiac Aging and Heart Failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Kajstura, J.; Pertoldi, B.; Leri, A.; Beltrami, C.-A.; Deptala, A.; Darzynkiewicz, Z.; Anversa, P. Telomere Shortening Is an in Vivo Marker of Myocyte Replication and Aging. Am. J. Pathol. 2000, 156, 813–819. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.A. A Time to Press Reset and Regenerate Cardiac Stem Cell Biology. JAMA Cardiol. 2019, 4, 95–96. [Google Scholar] [CrossRef]
- Shi, J.; Sun, J.; Liu, L.; Shan, T.; Meng, H.; Yang, T.; Wang, S.; Wei, T.; Chen, B.; Ma, Y.; et al. P16ink4a overexpression ameliorates cardiac remodeling of mouse following myocardial infarction via CDK4/pRb pathway. Biochem. Biophys. Res. Commun. 2022, 595, 62–68. [Google Scholar] [CrossRef]
- Joosten, S.A.; van Ham, V.; Nolan, C.E.; Borrias, M.C.; Jardine, A.G.; Shiels, P.G.; van Kooten, C.; Paul, L.C. Telomere Shortening and Cellular Senescence in a Model of Chronic Renal Allograft Rejection. Am. J. Pathol. 2003, 162, 1305–1312. [Google Scholar] [CrossRef]
- Chkhotua, A.B.; Gabusi, E.; Altimari, A.; D’Errico, A.; Yakubovich, M.; Vienken, J.; Stefoni, S.; Chieco, P.; Yussim, A.; Grigioni, W.F. Increased expression of p16(INK4a) and p27(Kip1) cyclin-dependent kinase inhibitor genes in aging human kidney and chronic allograft nephropathy. Am. J. Kidney Dis. 2003, 41, 1303–1313. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.W.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased Expression of Senescence-Associated Cell Cycle Inhibitor p16INK4a in Deteriorating Renal Transplants and Diseased Native Kidney. Am. J. Transplant. 2005, 5, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef]
- Sis, B.; Tasanarong, A.; Khoshjou, F.; Dadras, F.; Solez, K.; Halloran, P.F. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007, 71, 218–226. [Google Scholar] [CrossRef]
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.-R.; Chen, X.-M.; Cai, G.-Y.; Lin, L.-R.; He, Y.-N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol.-Cell Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef]
- Gu, X.; Peng, C.-Y.; Lin, S.-Y.; Qin, Z.-Y.; Liang, J.-L.; Chen, H.-J.; Hou, C.-X.; Wang, R.; Du, Y.-Q.; Jin, J.-L.; et al. P16INK4a played a critical role in exacerbating acute tubular necrosis in acute kidney injury. Am. J. Transl. Res. 2019, 11, 3850–3861. [Google Scholar]
- Jin, J.; Tao, J.; Gu, X.; Yu, Z.; Wang, R.; Zuo, G.; Li, Q.; Lv, X.; Miao, D. P16 INK4a Deletion Ameliorated Renal Tubulointerstitial Injury in a Stress-induced Premature Senescence Model of Bmi-1 Deficiency. Sci. Rep. 2017, 7, 7502. [Google Scholar] [CrossRef]
- Baiocchi, L.; Glaser, S.; Francis, H.; Kennedy, L.; Felli, E.; Alpini, G.; Gracia-Sancho, J. Impact of Aging on Liver Cells and Liver Disease: Focus on the Biliary and Vascular Compartments. Hepatol. Commun. 2021, 5, 1125–1137. [Google Scholar] [CrossRef]
- Kim, H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ikemoto, T.; Utsunomiya, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Shimada, M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J. Gastroenterol. Hepatol. 2014, 29, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N. Hepatocytes from old rats retain responsiveness of c-myc expression to EGF in primary culture but do not enter S phase. Exp. Cell Res. 1989, 181, 584–588. [Google Scholar] [CrossRef]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Hide, D.; Muñoz, L.; Hessheimer, A.J.; Vila, S.; Francés, R.; Fondevila, C.; Albillos, A.; et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018, 17, e12829. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Wang, T.-W.; Johmura, Y.; Kanai, T.; Nakano, Y.; Kido, T.; Susaki, E.A.; Nakajima, T.; Shichino, S.; Ueha, S.; et al. Generation of a p16 Reporter Mouse and Its Use to Characterize and Target p16high Cells In Vivo. Cell Metab. 2020, 32, 814–828e6. [Google Scholar] [CrossRef]
- González-Navarro, H.; Vinué, Á.; Sanz, M.J.; Delgado, M.; Pozo, M.A.; Serrano, M.; Burks, D.J.; Andrés, V. Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with aging. Aging Cell 2013, 12, 102–111. [Google Scholar] [CrossRef]
- Pal, A.; Potjer, T.P.; Thomsen, S.K.; Ng, H.J.; Barrett, A.; Scharfmann, R.; James, T.J.; Bishop, D.T.; Karpe, F.; Godsland, I.F.; et al. Loss-of-Function Mutations in the Cell-Cycle Control Gene CDKN2A Impact on Glucose Homeostasis in Humans. Diabetes 2016, 65, 527–533. [Google Scholar] [CrossRef]
- Bantubungi, K.; Hannou, S.-A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1α pathway. Diabetes 2014, 63, 3199–3209. [Google Scholar] [CrossRef][Green Version]
- Deleye, Y.; Cotte, A.K.; Hannou, S.A.; Hennuyer, N.; Bernard, L.; Derudas, B.; Caron, S.; Legry, V.; Vallez, E.; Dorchies, E.; et al. CDKN2A/p16INK4a suppresses hepatic fatty acid oxidation through the AMPKα2-SIRT1-PPARα signaling pathway. J. Biol. Chem. 2020, 295, 17310–17322. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, G.B.; Zhou, D.; Pan, Y.-X. High-fat diet modifies expression of hepatic cellular senescence gene p16(INK4a) through chromatin modifications in adult male rats. Genes Nutr. 2018, 13, 6. [Google Scholar] [CrossRef]
- Kyritsi, K.; Francis, H.; Zhou, T.; Ceci, L.; Wu, N.; Yang, Z.; Meng, F.; Chen, L.; Baiocchi, L.; Kundu, D.; et al. Downregulation of p16 Decreases Biliary Damage and Liver Fibrosis in the Mdr2−/− Mouse Model of Primary Sclerosing Cholangitis. Gene Expr. 2020, 20, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Li, N.; Kong, M.; Wu, J.; Fan, Z.; Miao, D.; Xu, Y.; Ye, Q.; Wang, Y. CDKN2a/p16 Antagonizes Hepatic Stellate Cell Activation and Liver Fibrosis by Modulating ROS Levels. Front. Cell Dev. Biol. 2020, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.-J.; Xie, F.; Xu, J.-F.; Qin, Y.-Y.; Shen, R.-X.; Yang, J.-M.; He, J. P16 gene hypermethylation and hepatocellular carcinoma: A systematic review and meta-analysis. World J. Gastroenterol. 2011, 17, 3043–3048. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.H.N.; Dennis Lo, Y.M.; Zhang, J.; Liew, C.-T.; Ng, M.H.L.; Wong, N.; Lai, P.B.S.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of Aberrant p16 Methylation in the Plasma and Serum of Liver Cancer Patients1. Cancer Res. 1999, 59, 71–73. [Google Scholar] [PubMed]
- Wagner, K.-D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and p21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef] [PubMed]
- Vasey, D.B.; Roland Wolf, C.; Brown, K.; Whitelaw, C.B.A. Spatial p21 expression profile in the mid-term mouse embryo. Transgenic Res. 2011, 20, 23–28. [Google Scholar] [CrossRef]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.W.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003, 63, 2134–2143. [Google Scholar] [CrossRef]
- Rhinn, M.; Zapata-Bodalo, I.; Klein, A.; Plassat, J.-L.; Knauer-Meyer, T.; Keyes, W.M. Aberrant induction of p19Arf-mediated cellular senescence contributes to neurodevelopmental defects. PLoS Biol. 2022, 20, e3001664. [Google Scholar] [CrossRef]
- Cheong, C.; Sung, Y.H.; Lee, J.; Choi, Y.S.; Song, J.; Kee, C.; Lee, H.-W. Role of INK4a locus in normal eye development and cataract genesis. Mech. Ageing Dev. 2006, 127, 633–638. [Google Scholar] [CrossRef]
- Nacher, V.; Carretero, A.; Navarro, M.; Ayuso, E.; Ramos, D.; Luppo, M.; Rodríguez, A.; Mendes, L.; Herrero-Fresneda, I.; Ruberte, J. Endothelial Cell Transduction in Primary Cultures from Regressing Mesonephros. Cells Tissues Organs 2010, 191, 84–95. [Google Scholar] [CrossRef]
- Domínguez-Bautista, J.A.; Acevo-Rodríguez, P.S.; Castro-Obregón, S. Programmed Cell Senescence in the Mouse Developing Spinal Cord and Notochord. Front. Cell Dev. Biol. 2021, 9, 587096. [Google Scholar] [CrossRef]
- Wagner, N.; Ninkov, M.; Vukolic, A.; Cubukcuoglu Deniz, G.; Rassoulzadegan, M.; Michiels, J.-F.; Wagner, K.-D. Implications of the Wilms’ Tumor Suppressor Wt1 in Cardiomyocyte Differentiation. Int. J. Mol. Sci. 2021, 22, 94346. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.-F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef]
- El Maï, M.; Wagner, K.-D.; Michiels, J.-F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.-J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef]
- Wagner, K.-D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.-F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Morrison, H.; Pagnotta, S.; Michiels, J.-F.; Schwab, Y.; Tryggvason, K.; Schedl, A.; Wagner, K.-D. The podocyte protein nephrin is required for cardiac vessel formation. Hum. Mol. Genet. 2011, 20, 2182–2194. [Google Scholar] [CrossRef]
- Dimri, G.P. The search for biomarkers of aging: Next stop INK4a/ARF locus. Sci. Aging Knowl. Environ. 2004, 2004, pe40. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Hall, B.M.; Strom, E.; Virtuoso, L.P.; Gupta, M.; Gleiberman, A.S.; Rydkina, E.; Balan, V.; Vujcic, S.; Chernova, O.B.; et al. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle 2017, 16, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Misra, A.; Netto, J.M.E.; Tchkonia, T.; Kirkland, J.L. Strategies for late phase preclinical and early clinical trials of senolytics. Mech. Ageing Dev. 2021, 200, 111591. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safwan-Zaiter, H.; Wagner, N.; Wagner, K.-D. P16INK4A—More Than a Senescence Marker. Life 2022, 12, 1332. https://doi.org/10.3390/life12091332
Safwan-Zaiter H, Wagner N, Wagner K-D. P16INK4A—More Than a Senescence Marker. Life. 2022; 12(9):1332. https://doi.org/10.3390/life12091332
Chicago/Turabian StyleSafwan-Zaiter, Hasan, Nicole Wagner, and Kay-Dietrich Wagner. 2022. "P16INK4A—More Than a Senescence Marker" Life 12, no. 9: 1332. https://doi.org/10.3390/life12091332
APA StyleSafwan-Zaiter, H., Wagner, N., & Wagner, K.-D. (2022). P16INK4A—More Than a Senescence Marker. Life, 12(9), 1332. https://doi.org/10.3390/life12091332