
Protective Effects of High-Fat Diet against Murine Colitis in Association with Leptin Signaling and Gut Microbiome

 , , ,
, , ,  and
and 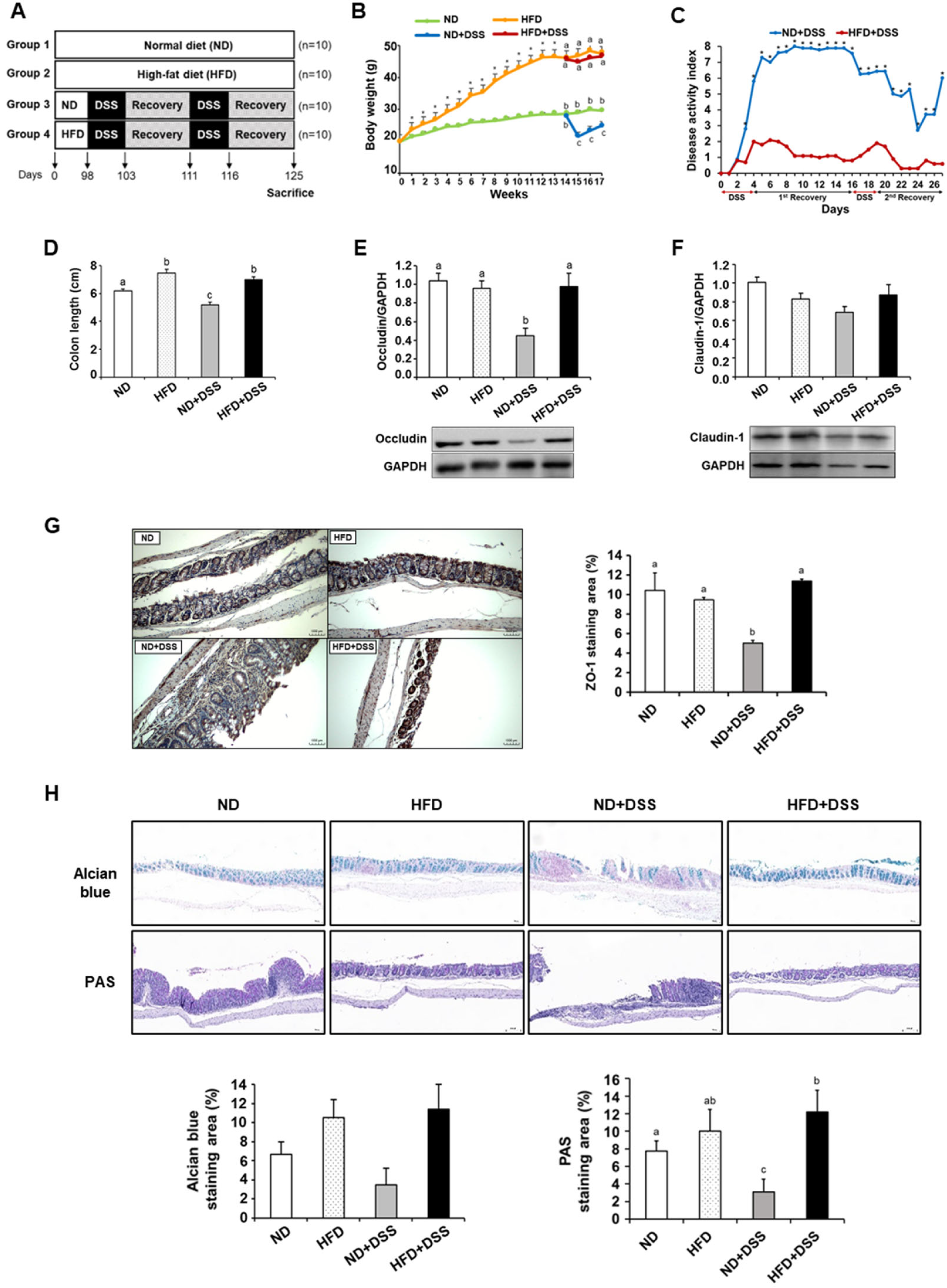{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Histological and Immunohistochemical Analyses
2.3. Flow Cytometry
2.4. Measurement of Serum Leptin Concentration
2.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Western Blot Analysis
2.7. Pyrosequencing
2.8. Statistical Analysis
3. Results
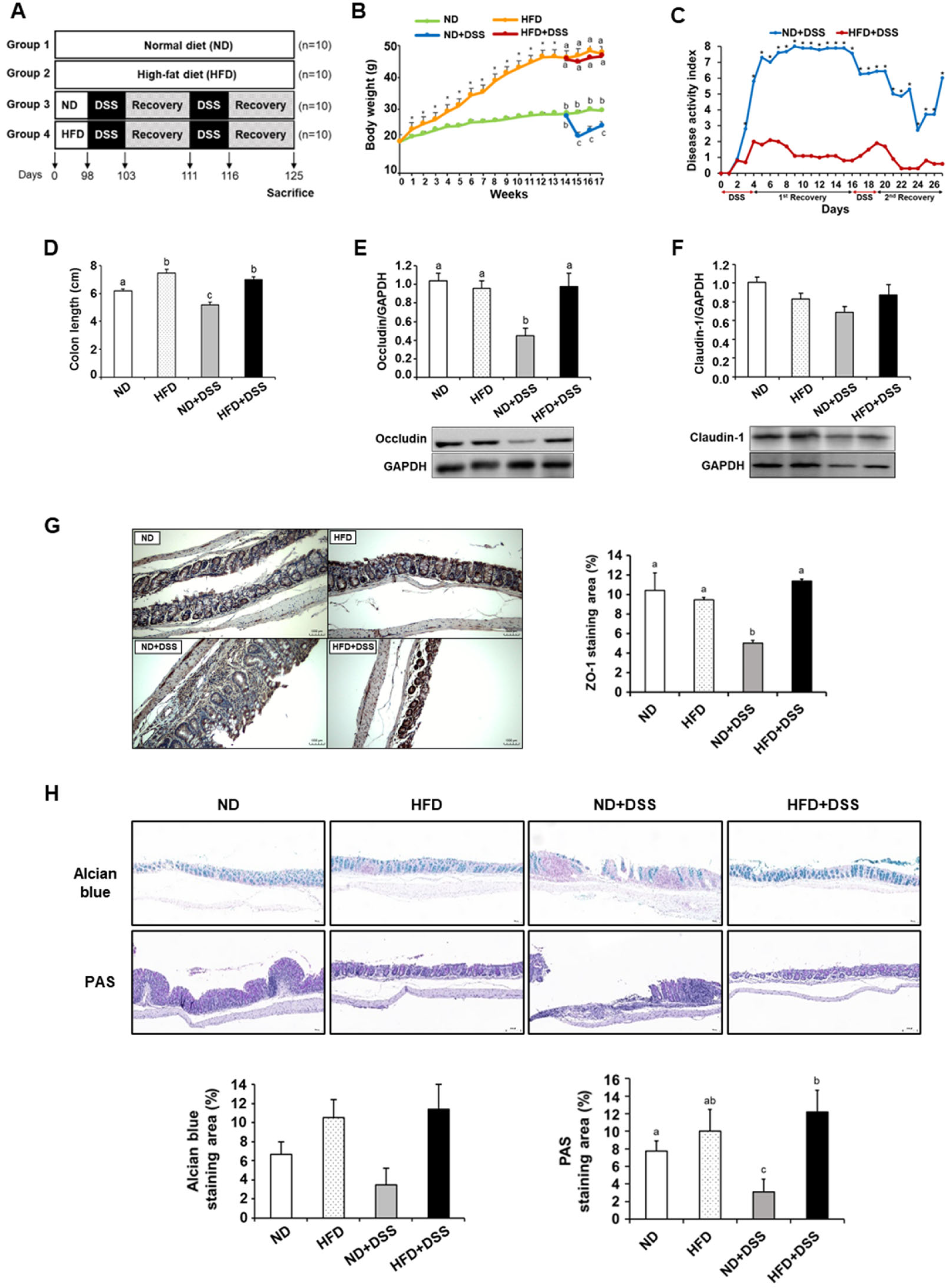
3.1. HFD Feeding Alleviates DSS-Induced Colitis Symptoms and Reinforces Colonic Barrier Function
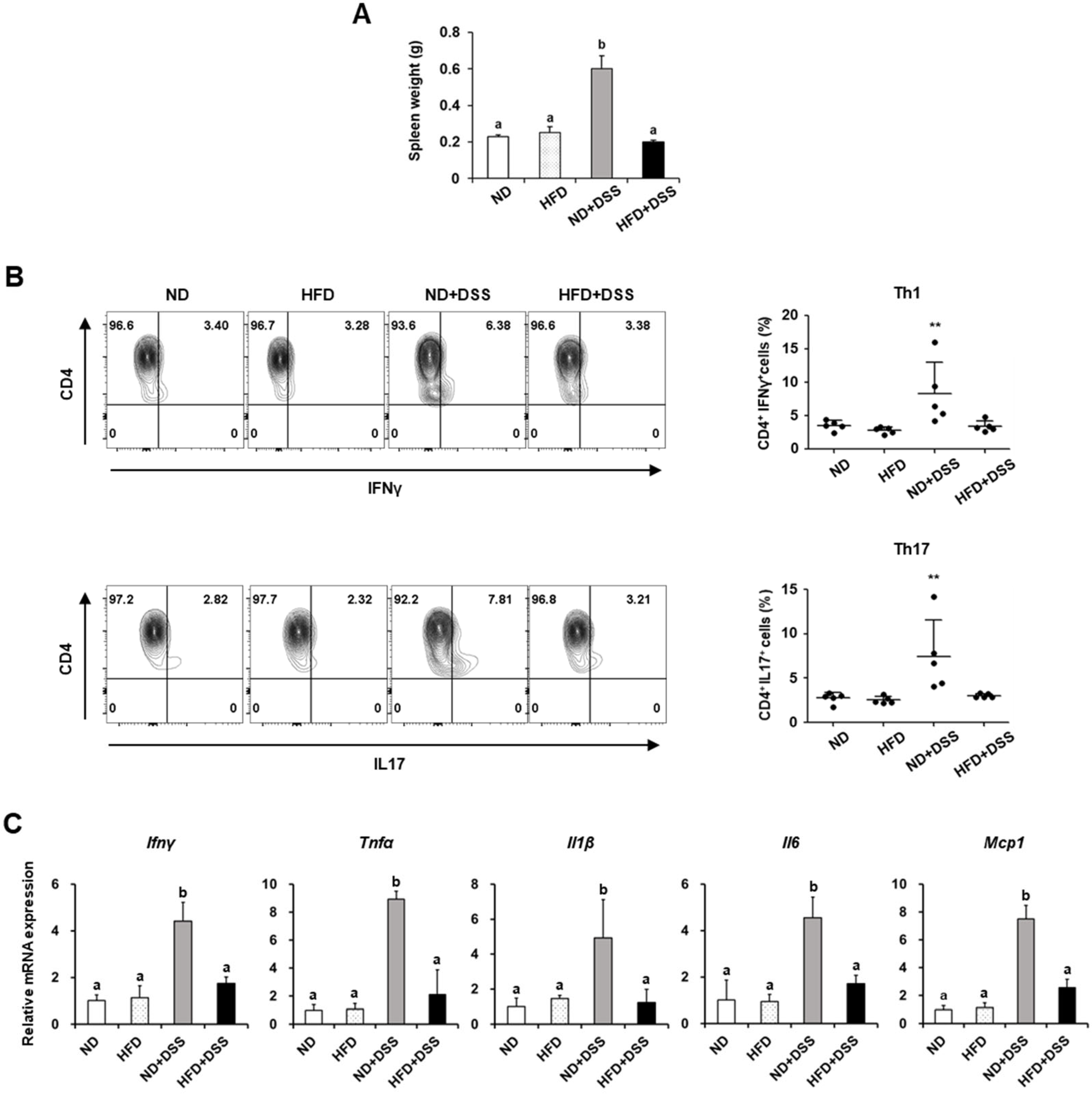
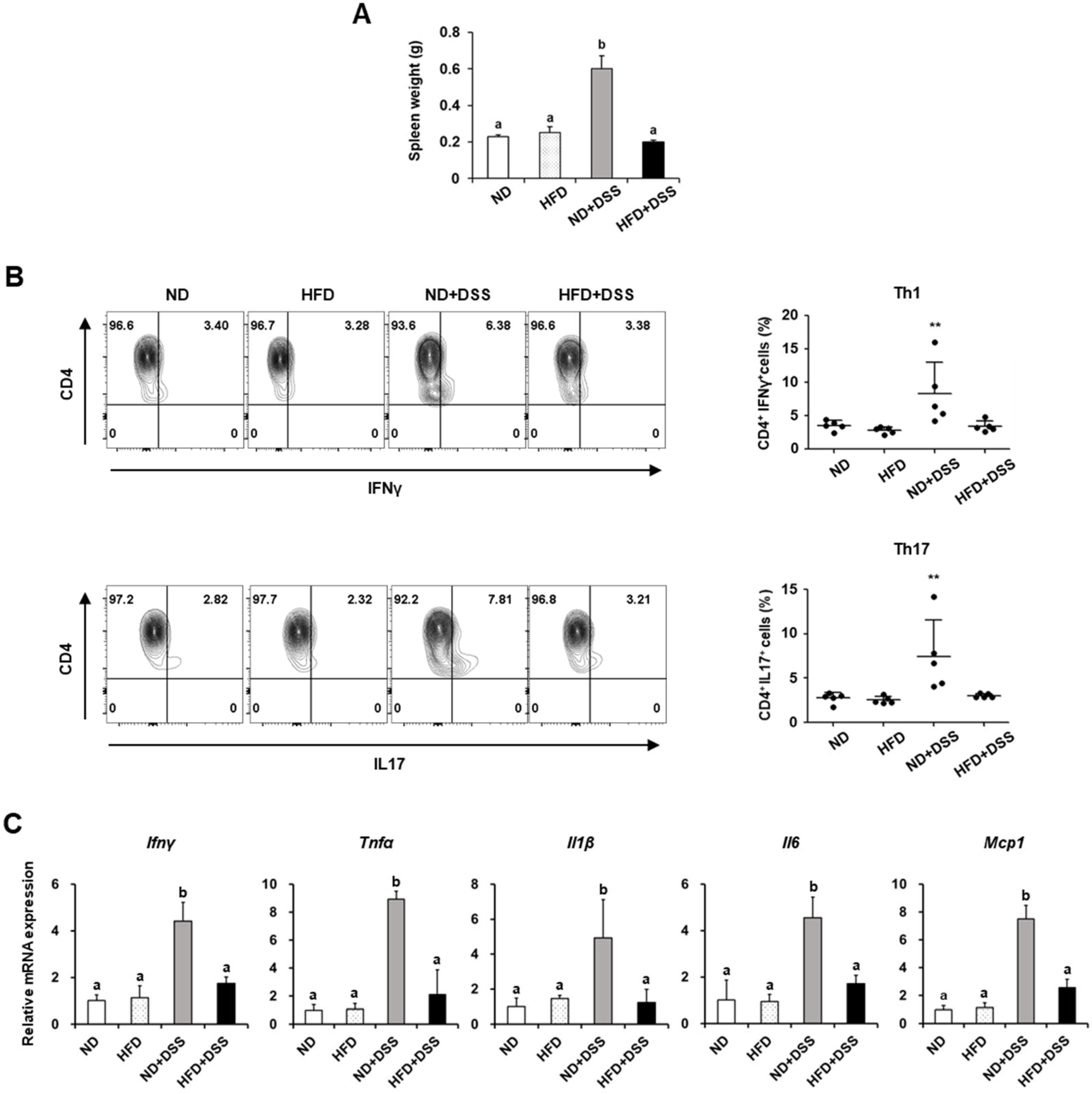
3.2. HFD Feeding Improves Immune Responses in DSS-Induced Colitis
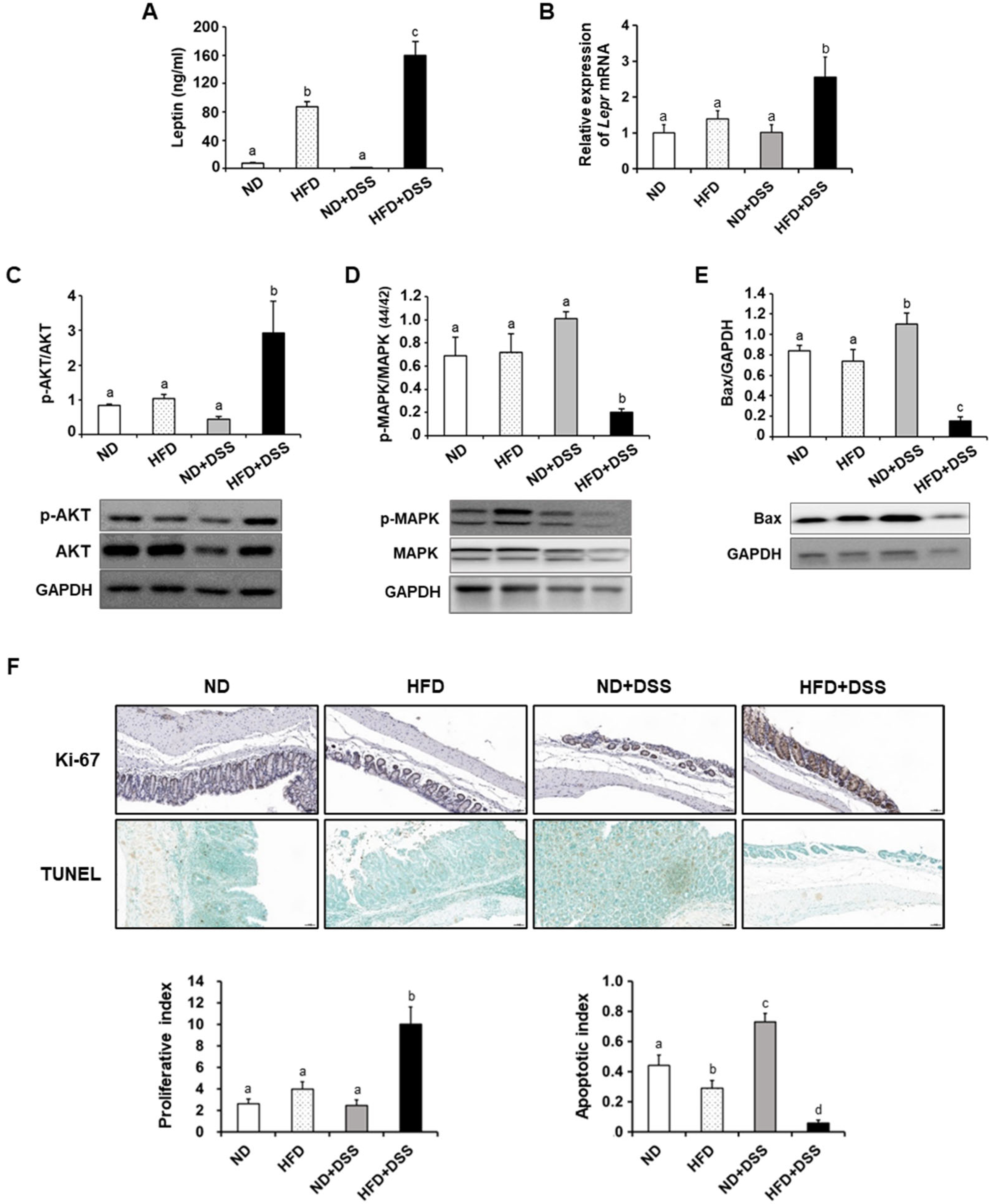
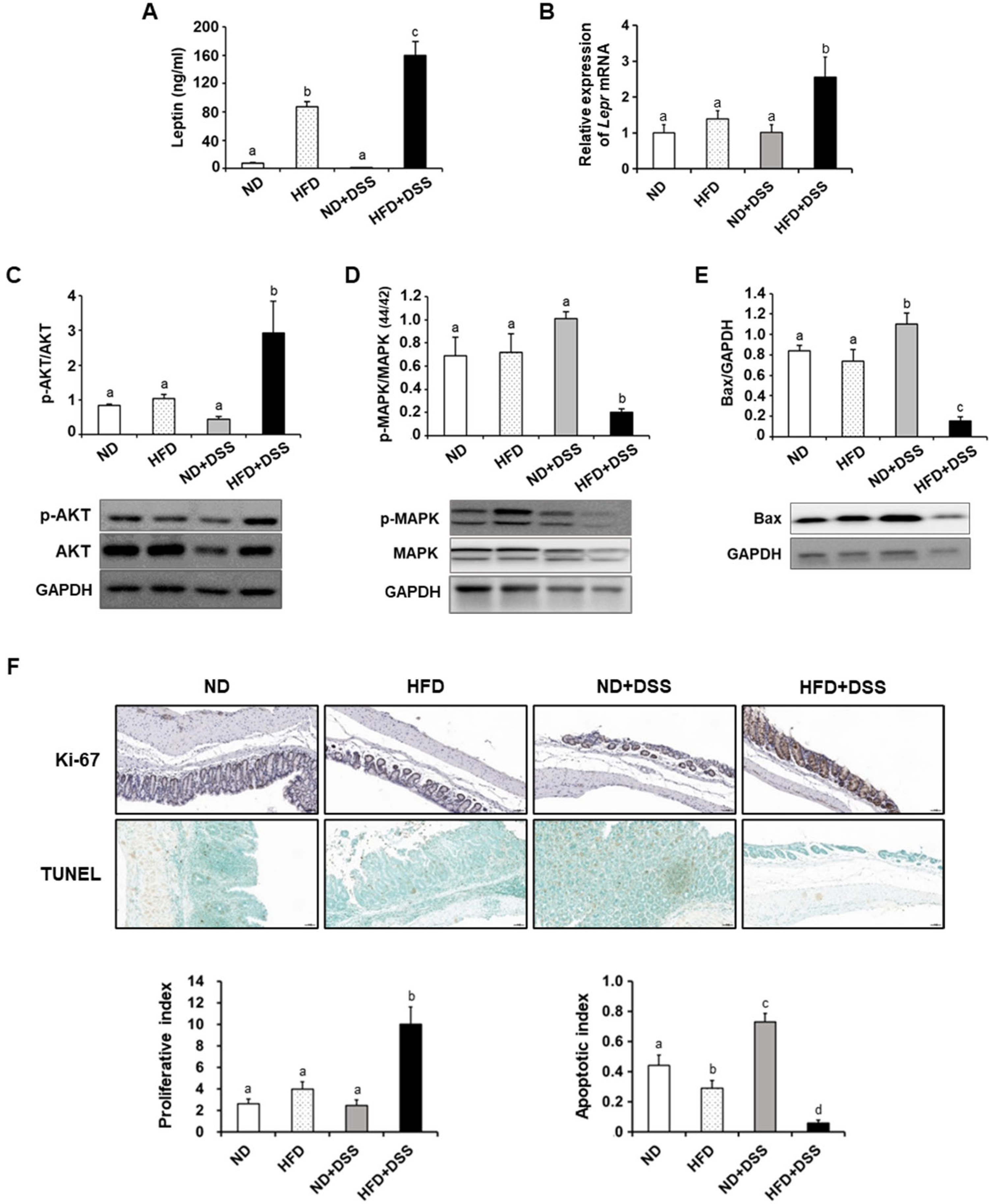
3.3. HFD Feeding Regulates Apoptosis-Associated Molecules in the Inflamed Intestinal Epithelium
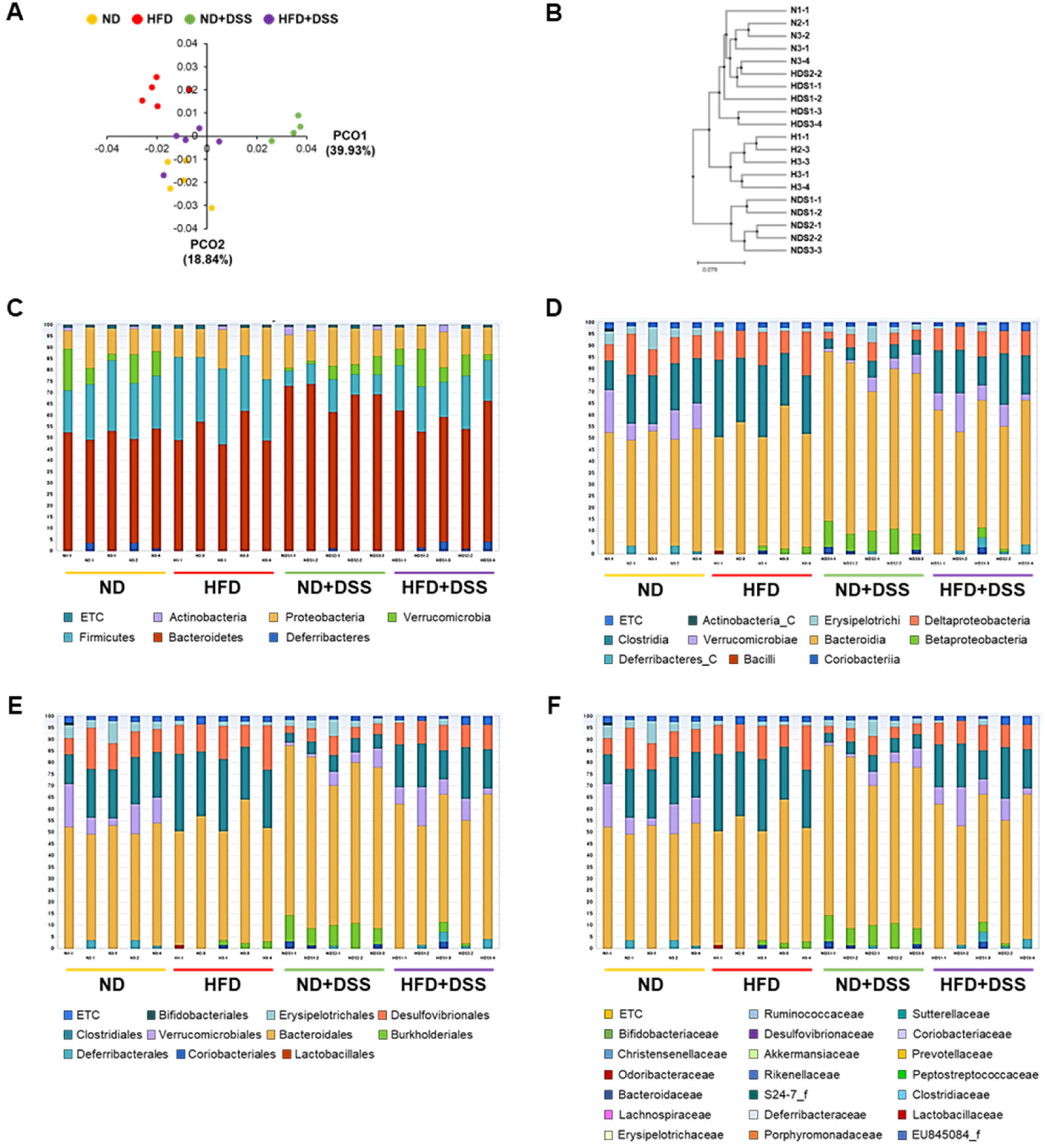
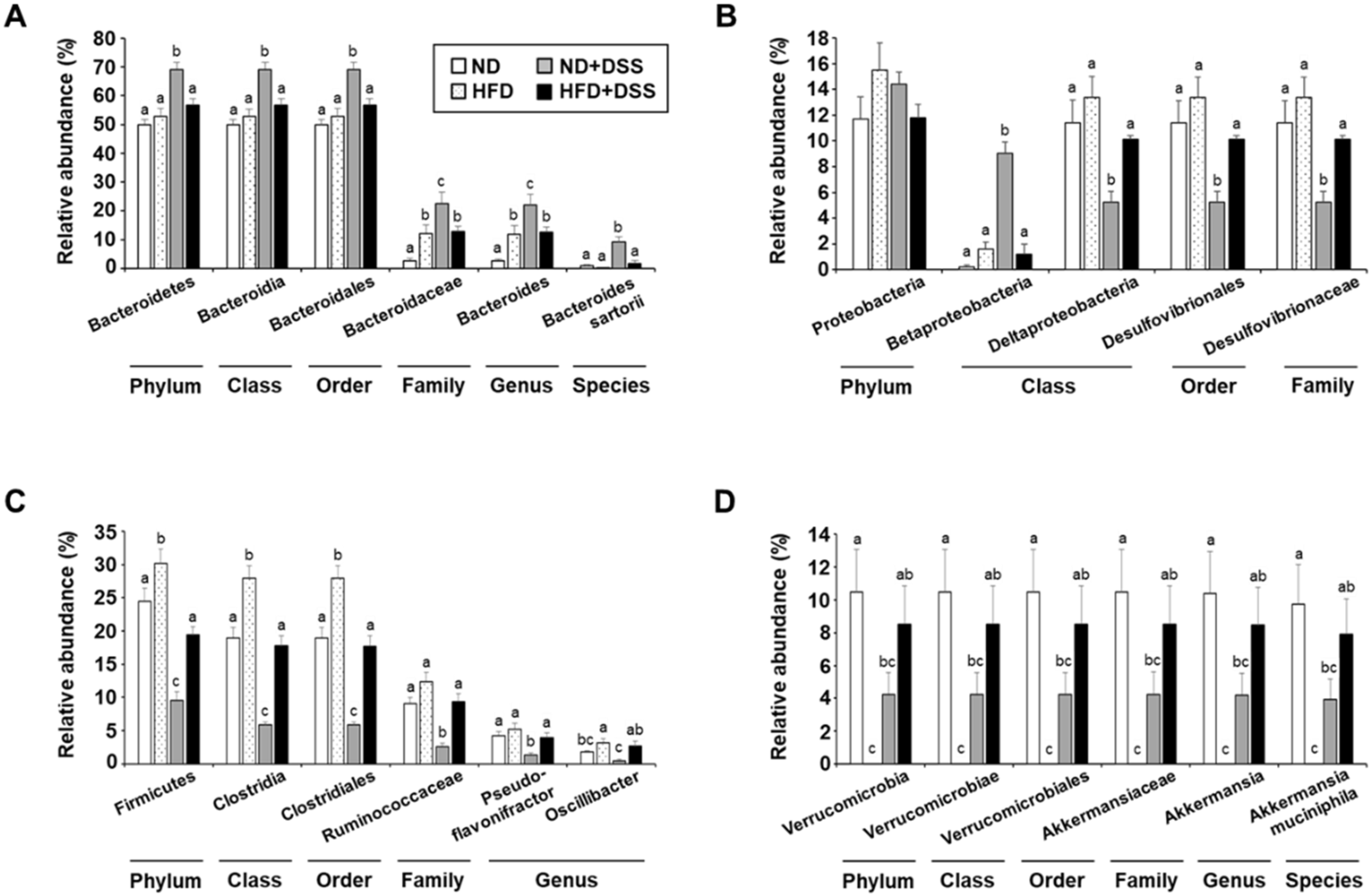
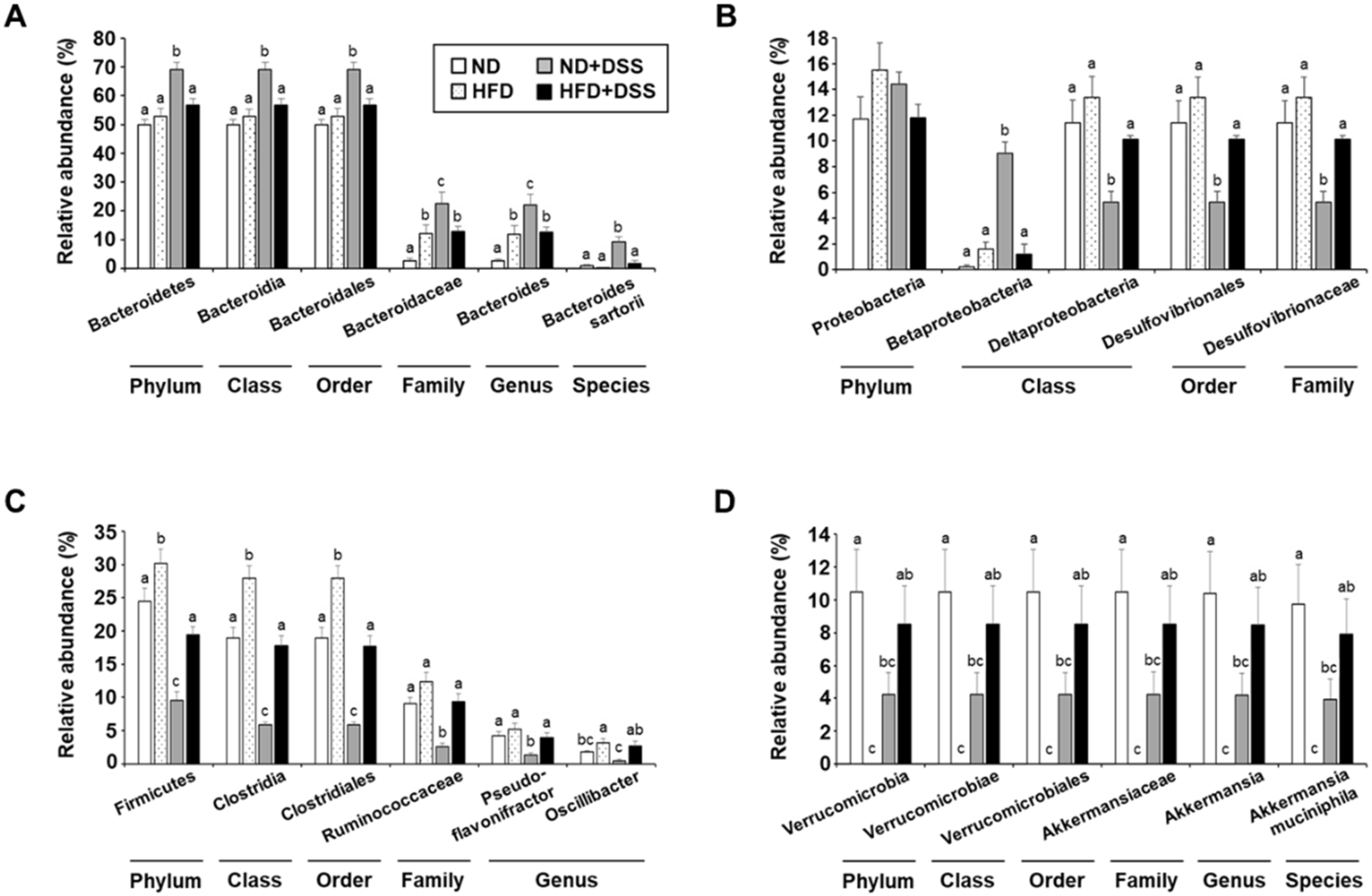
3.4. HFD Feeding Induces Alterations in Gut Microbiota Diversity and Taxonomic Composition
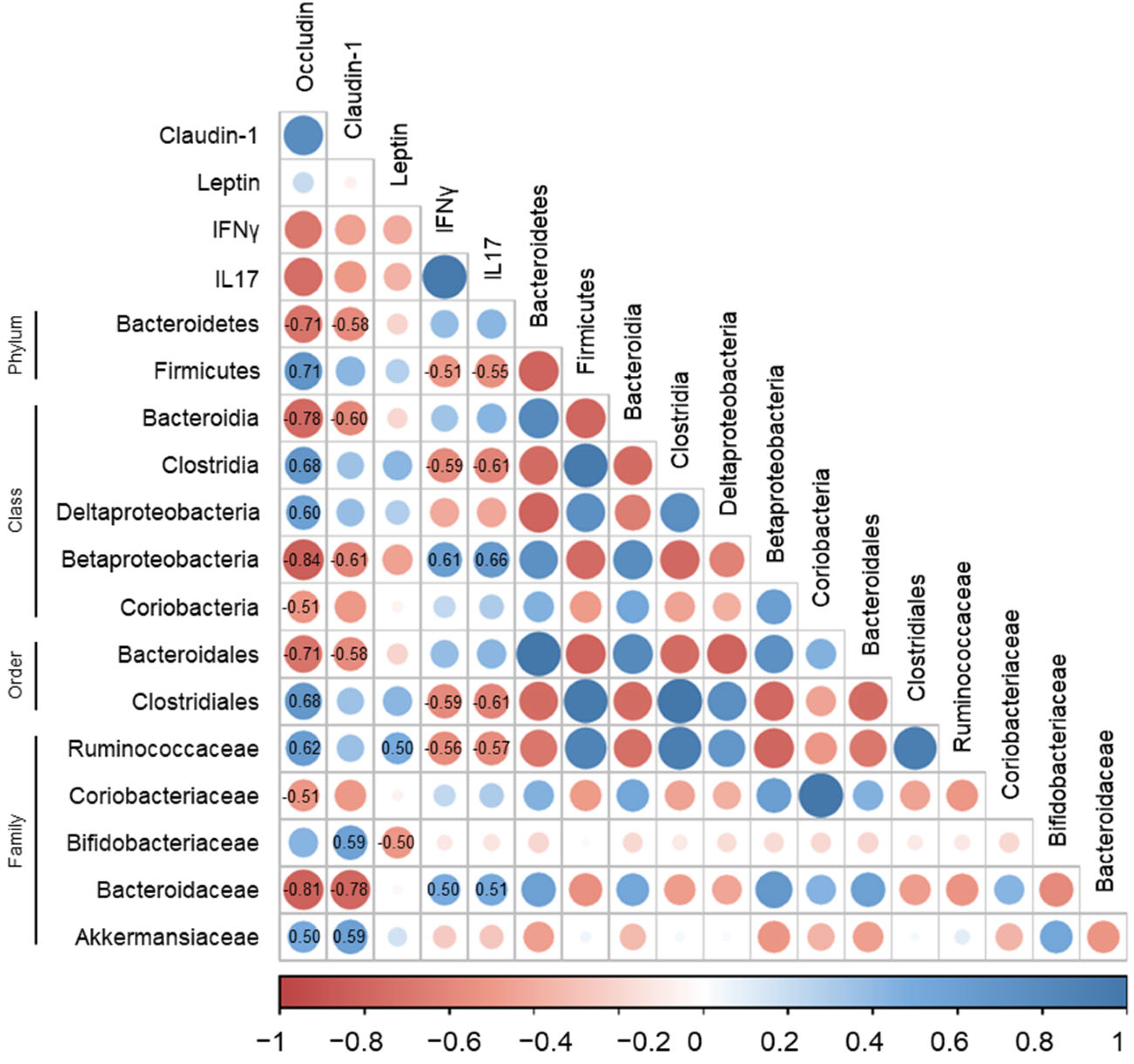
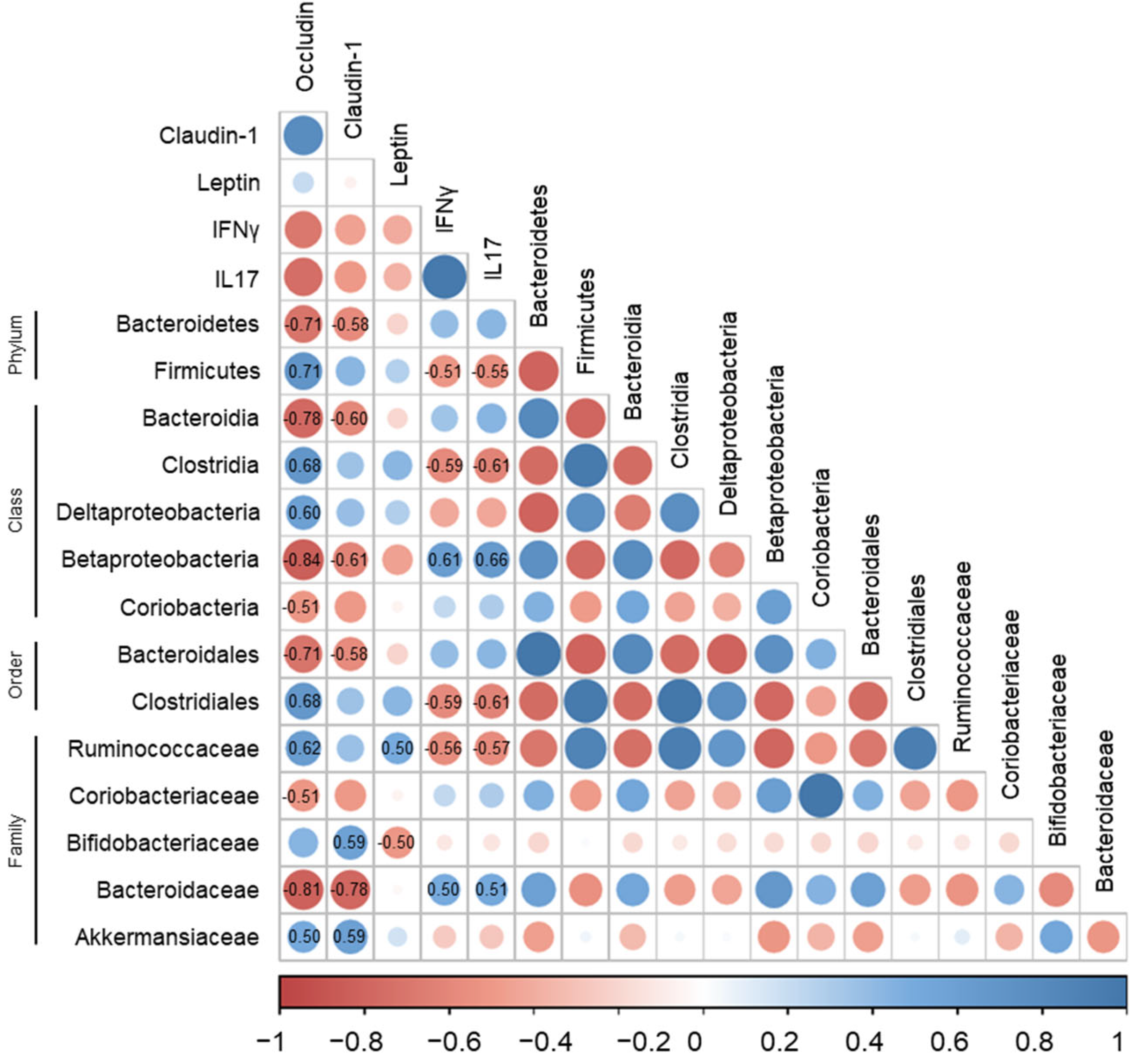
3.5. Correlations of Gut Microbiota with Immune-Response Markers and Tight-Junction Proteins in DSS-Induced Colitis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; MacPherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 39–49. [Google Scholar] [CrossRef]
- Elsherif, Y.; Alexakis, C.; Mendall, M. Determinants of Weight Loss prior to Diagnosis in Inflammatory Bowel Disease: A Retrospective Observational Study. Gastroenterol. Res. Pract. 2014, 2014, 762191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Chen, Y.; Tang, Y.; Xu, F.; Yu, C.; Li, Y.; Pankaj, P.; Dai, N. Body Mass Index Is Associated with Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0144872. [Google Scholar] [CrossRef] [Green Version]
- Rizzello, F.; Spisni, E.; Giovanardi, E.; Imbesi, V.; Salice, M.; Alvisi, P.; Valerii, M.C.; Gionchetti, P. Implications of the Westernized Diet in the Onset and Progression of IBD. Nutrients 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.; Boneh, R.S.; Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 2018, 67, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.F.; Costanzo, A.; Gareau, M.G.; Armando, A.M.; Quehenberger, O.; Jameson, J.M.; Olefsky, J.M. High Fat Diet Causes Depletion of Intestinal Eosinophils Associated with Intestinal Permeability. PLoS ONE 2015, 10, e0122195. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Jin, H.; Qiang, Y.; Wu, S.; Yan, C.; Han, M.; Xiao, T.; Yan, N.; An, H.; Zhou, X.; et al. High fat diet exacerbates dextran sulfate sodium induced colitis through disturbing mucosal dendritic cell homeostasis. Int. Immunopharmacol. 2016, 40, 1–10. [Google Scholar] [CrossRef]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2013, 63, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Lin, X.; Zhao, Q.; Li, J. Fat intake and risk of ulcerative colitis: Systematic review and dose-response meta-analysis of epidemiological studies. J. Gastroenterol. Hepatol. 2017, 32, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Mackey-Lawrence, N.M.; Petri, W.A., Jr. Leptin and mucosal immunity. Mucosal Immunol. 2012, 5, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, A.P.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Vazquez, F.; Garza-Veloz, I.; Villela-Ramirez, G.A.; Ortiz-Castro, Y.; Mauricio-Saucedo, P.; Cardenas-Vargas, E.; Di-az-Baez, M.; Cid-Baez, M.A.; Castañeda-Miranda, R.; Ortiz-Rodriguez, J.M. Positive association between leptin serum levels and disease activity on endoscopy in inflammatory bowel disease: A case-control study. Exp. Ther. Med. 2018, 15, 3336–3344. [Google Scholar] [CrossRef] [Green Version]
- Karmiris, K.; Koutroubakis, I.; Xidakis, C.; Polychronaki, M.; Voudouri, T.; Kouroumalis, E.A. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 100–105. [Google Scholar] [CrossRef]
- Waluga, M.; Hartleb, M.; Boryczka, G.; Kukla, M.; Żwirska-Korczala, K. Serum adipokines in inflammatory bowel disease. World J. Gastroenterol. WJG 2014, 20, 6912. [Google Scholar] [CrossRef]
- Albenberg, L.G.; Wu, G.D. Diet and the Intestinal Microbiome: Associations, Functions, and Implications for Health and Disease. Gastroenterology 2014, 146, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.; Gulati, A.; Sartor, R.B. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol. 2010, 26, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill–Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.-Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-Fat Diet Determines the Composition of the Murine Gut Microbiome Independently of Obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Nam, S.; Kang, K.; Cha, J.S.; Kim, J.W.; Lee, H.G.; Kim, Y.; Yang, Y.; Lee, M.-S.; Lim, J.-S. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J. Leukoc. Biol. 2016, 100, 1273–1284. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M. Role of Goblet Cells in Intestinal Barrier and Mucosal Immunity. J. Inflamm. Res. 2021, 14, 3171–3183. [Google Scholar] [CrossRef]
- Clayburgh, D.; Shen, L.; Turner, J.R. A porous defense: The leaky epithelial barrier in intestinal disease. Lab. Investig. 2004, 84, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Bruewer, M.; Samarin, S.; Nusrat, A. Inflammatory Bowel Disease and the Apical Junctional Complex. Ann. Acad. Sci. 2006, 1072, 242–252. [Google Scholar] [CrossRef]
- Weber, C.; Turner, J. Inflammatory bowel disease: Is it really just another break in the wall? Gut 2007, 56, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Pittet, M.J. The Spleen in Local and Systemic Regulation of Immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Markel, J.E.; Noore, J.; Emery, E.J.; Bobnar, H.J.; Kleinerman, E.S.; Lindsey, B.A. Using the Spleen as an In Vivo Systemic Immune Barometer Alongside Osteosarcoma Disease Progression and Immunotherapy with α-PD-L1. Sarcoma 2018, 2018, 8694397. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [Green Version]
- Günther, C.; Neumann, H.; Neurath, M.F.; Becker, C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut 2012, 62, 1062–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L.P. The anti-apoptotic and growth stimulatory actions of leptin in human colon cancer cells involves activation of JNK mitogen activated protein kinase, JAK2 and PI3 kinase/Akt. Int. J. Color. Dis. 2006, 22, 401–409. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, D.; Moran, C.; Shanahan, F. The microbiota in inflammatory bowel disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, T.; Aiba, Y.; Ishikawa, H.; Ushiyama, A.; Takagi, A.; Mine, T.; Koga, Y. The suppressive effect of bifidobacteria on Bacteroides vulgatus, a putative pathogenic microbe in inflammatory bowel disease. Microbiol. Immunol. 2003, 47, 371–378. [Google Scholar] [CrossRef]
- Sun, J. Pathogenic Bacterial Proteins and their Anti-Inflammatory Effects in the Eukaryotic Host. Anti-Inflammat Anti-Allergy Agents Med. Chem. 2009, 8, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Håkansson, Å.; Tormobadia, N.; Baridi, A.; Xu, J.; Molin, G.; Hagslätt, M.-L.; Karlsson, C.; Jeppsson, B.; Cilio, C.M.; Ahrné, S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. 2015, 15, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, L.G.; Leonel, A.J.; Aguilar, E.C.; Batista, N.V.; Alves, A.C.; Coimbra, C.C.; Ferreira, A.V.; de Faria, A.M.C.; Cara, D.C.; Alvarez Leite, J.I. The combination of high-fat diet-induced obesity and chronic ulcerative colitis reciprocally exacerbates adipose tissue and colon inflammation. Lipids Health Dis. 2011, 10, 204. [Google Scholar] [CrossRef] [Green Version]
- El Homsi, M.; Ducroc, R.; Claustre, J.; Jourdan, G.; Gertler, A.; Estienne, M.; Bado, A.; Scoazec, J.-Y.; Plaisancié, P. Leptin modulates the expression of secreted and membrane-associated mucins in colonic epithelial cells by targeting PKC, PI3K, and MAPK pathways. Am. J. Physiol. Liver Physiol. 2007, 293, G365–G373. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Roberts, M.R.; Becker, S.M.; Podd, B.; Zhang, Y.; Chua, S.C., Jr.; Myers, M.G., Jr.; Duggal, P.; Houpt, E.R.; Petri, W.A., Jr. Leptin signaling in intestinal epithelium mediates resistance to enteric infection by Entamoeba histolytica. Mucosal Immunol. 2011, 4, 294. [Google Scholar] [CrossRef] [Green Version]
- Van der Sluis, M.; de Koning, B.A.E.; de Bruijn, A.C.J.M.; Velcich, A.; Meijerink, J.P.P.; van Goudoever, J.B.; Büller, H.A.; Dekker, J.; VAN Seuningen, I.; Renes, I.B.; et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That MUC2 Is Critical for Colonic Protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Petersson, J.; Schreiber, O.; Hansson, G.C.; Gendler, S.J.; Velcich, A.; Lundberg, J.O.; Roos, S.; Holm, L.; Phillipson, M. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am. J. Physiol. Liver Physiol. 2011, 300, G327–G333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chami, B.; Yeung, A.W.S.; van Vreden, C.; King, N.J.C.; Bao, S. The Role of CXCR3 in DSS-Induced Colitis. PLoS ONE 2014, 9, e101622. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-P.; Yuan, H.; Xu, Y.; Liu, R.-M.; Luo, Y.; Xiao, J.-H. Protective effects of Ligularia fischeri root extracts against ulcerative colitis in mice through activation of Bcl-2/Bax signalings. Phytomedicine 2022, 99, 154006. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.Y.; Moon, J.; Choi, J.; Jhun, J.; Jung, K.; Cho, K.-H.; Darlami, O.; Lee, H.H.; Jung, E.S.; et al. Inhibition of RIPK3 Pathway Attenuates Intestinal Inflammation and Cell Death of Inflammatory Bowel Disease and Suppresses Necroptosis in Peripheral Mononuclear Cells of Ulcerative Colitis Patients. Immune Netw. 2020, 20, e16. [Google Scholar] [CrossRef]
- Chi, J.H.; Kim, Y.H.; Sohn, D.H.; Seo, G.S.; Lee, S.H. Ameliorative effect of Alnus japonica ethanol extract on colitis through the inhibition of inflammatory responses and attenuation of intestinal barrier disruption in vivo and in vitro. Biomed. Pharmacother. 2018, 108, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- De La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr. Metab. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Chen, X.; Kolaczynski, J.W.; Polansky, M. Effects of prolonged hyperinsulinemia on serum leptin in normal human subjects. J. Clin. Investig. 1997, 100, 1107–1113. [Google Scholar] [CrossRef]
- Zakrzewska, K.E.; Cusin, I.; Sainsbury, A.; Rohner-Jeanrenaud, F.; Jeanrenaud, B. Glucocorticoids as counterregulatory hormones of leptin: Toward an understanding of leptin resistance. Diabetes 1997, 46, 717–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballinger, A.; Kelly, P.; Hallyburton, E.; Besser, R.; Farthing, M. Plasma Leptin in Chronic Inflammatory Bowel Disease and HIV: Implications for the Pathogenesis of Anorexia and Weight Loss. Clin. Sci. 1998, 94, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Hoppin, A.G.; Kaplan, L.M.; Zurakowski, D.; Leichtner, A.M.; Bousvaros, A. Serum Leptin in Children and Young Adults with Inflammatory Bowel Disease. J. Pediatr. Gastroenterol. Nutr. 1998, 26, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Merigo, F.; Brandolese, A.; Facchin, S.; Boschi, F.; Di Chio, M.; Savarino, E.; D’Incà, R.; Sturniolo, G.C.; Sbarbati, A. Immunolocalization of leptin and leptin receptor in colorectal mucosa of ulcerative colitis, Crohn’s disease and control subjects with no inflammatory bowel disease. Cell Tissue Res. 2020, 383, 1103–1122. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.G.F.; Lima, W.G.; Coelho, L.G.V.; Cardoso, V.N.; Fernandes, S.O.A. Circulating Leptin Levels as a Potential Biomarker in Inflammatory Bowel Diseases: A Systematic Review and Meta-Analysis. Inflamm. Bowel Dis. 2020, 27, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Plaisancie, P.; Ducroc, R.; Homsi, M.E.; Tsocas, A.; Guilmeau, S.; Zoghbi, S.; Thibaudeau, O.; Bado, A. Luminal leptin acti-vates mucin-secreting goblet cells in the large bowel. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G805–G812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Ren, X.; Jurickova, I.; Groschwitz, K.; Pasternak, B.A.; Xu, H.; Wilson, T.; Hogan, S.; Denson, L. Regulation of in-testinal barrier function by signal transducer and activator of transcription 5b. Gut 2009, 58, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Louis, N.A. Inflammatory Mediators Contributing to Intestinal Epithelial Cell Apoptosis and Barrier Disruption in IBD. J. Clin. Cell. Immunol. 2012, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.-H.; Yan, G.-T.; Wang, L.-H.; Zhang, J.-Y.; Xue, H.; Zhang, K. Leptin relieves intestinal ischemia/reperfusion injury by promoting ERK1/2 phosphorylation and the NO signaling pathway. J. Trauma: Inj. Infect. Crit. Care 2012, 72, 143–149. [Google Scholar] [CrossRef]
- Ye, C.; Wang, R.; Wang, M.; Huang, Z.; Tang, C. Leptin alleviates intestinal mucosal barrier injury and inflammation in obese mice with acute pancreatitis. Int. J. Obes. 2018, 42, 1471–1479. [Google Scholar] [CrossRef]
- Rivero-Gutiérrez, B.; Aranda, C.J.; Ocón, B.; Arredondo, M.; Martínez-Augustin, O.; de Medina, F.S. Exogenous leptin reinforces intestinal barrier function and protects from colitis. Pharmacol. Res. 2019, 147, 104356. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, S.; Guen, C.G.-L.; Le Vacon, F.; Potel, G.; de La Cochetiere, M.-F. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013, 21, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic Health: Fermentation and Short Chain Fatty Acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Peng, H.-W.; Wright, A.-D.G.; Bai, S.-P.; Ding, X.-M.; Zeng, Q.-F.; Li, H.; Zheng, P.; Su, Z.-W.; Cui, R.-Y.; et al. Broilers fed dietary vitamins harbor higher diversity of cecal bacteria and higher ratio of Clostridium, Faecalibacterium, and Lactobacillus than broilers with no dietary vitamins revealed by 16S rRNA gene clone libraries. Poult. Sci. 2013, 92, 2358–2366. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geerlings, S.Y.; Kostopoulos, I.; de Vos, W.M.; Belzer, C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Fujio-Vejar, S.; Vasquez, Y.; Morales, P.; Magne, F.; Vera-Wolf, P.; Ugalde, J.A.; Navarrete, P.; Gotteland, M. The gut mi-crobiota of healthy chilean subjects reveals a high abundance of the phylum verrucomicrobia. Front. Microbiol. 2017, 8, 1221. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-H.; Kim, H.; Nam, S.; Chu, J.-R.; Kim, J.-H.; Lim, J.-S.; Kim, S.-E.; Sung, M.-K. Protective Effects of High-Fat Diet against Murine Colitis in Association with Leptin Signaling and Gut Microbiome. Life 2022, 12, 972. https://doi.org/10.3390/life12070972
Lee Y-H, Kim H, Nam S, Chu J-R, Kim J-H, Lim J-S, Kim S-E, Sung M-K. Protective Effects of High-Fat Diet against Murine Colitis in Association with Leptin Signaling and Gut Microbiome. Life. 2022; 12(7):972. https://doi.org/10.3390/life12070972
Chicago/Turabian StyleLee, Yun-Ha, Hyeyoon Kim, Sorim Nam, Jae-Ryang Chu, Jung-Hwan Kim, Jong-Seok Lim, Sung-Eun Kim, and Mi-Kyung Sung. 2022. "Protective Effects of High-Fat Diet against Murine Colitis in Association with Leptin Signaling and Gut Microbiome" Life 12, no. 7: 972. https://doi.org/10.3390/life12070972
APA StyleLee, Y.-H., Kim, H., Nam, S., Chu, J.-R., Kim, J.-H., Lim, J.-S., Kim, S.-E., & Sung, M.-K. (2022). Protective Effects of High-Fat Diet against Murine Colitis in Association with Leptin Signaling and Gut Microbiome. Life, 12(7), 972. https://doi.org/10.3390/life12070972