1. Introduction
Cicadidae Periostracum (CP) is a crude drug derived from the cast-off skin of the insect Cryptotympana pustulata Fabricius from the family of Cicadas [
1,
2]. CP has been used to treat smallpox, sedation, shock, edema, epilepsy, and night terror symptoms in traditional Korean medicine. Various pharmacological studies on CP have shown its diverse biological properties, such as neuroprotective [
3]; immunomodulatory [
4]; anti-fibrosis [
1]; hypothermic, sedative, and anti-convulsive [
5]; anti-oxidative [
6]; and anti-inflammatory [
2,
4,
6] effects. CP is predominantly composed of proteins and chitin and a small number of minerals, lipids, and other constituents [
4,
7,
8]. Among the numerous components that make up a small proportion of CP, oleic acid (OA) accounts for a relatively large proportion of the total fatty acids in the ethanol extract of CP, which was previously reported by our group [
7]. OA is a monounsaturated and non-essential fatty acid and has demonstrated anti-inflammatory [
9,
10,
11,
12] and anti-tumor [
13] activities. Although the role of OA in immune responses is still controversial [
9,
10], previously, our group demonstrated the anti-asthmatic effects of CP and OA in OVA-induced asthmatic mouse models [
7].
Allergic asthma is a chronic airway disease that involves complex interactions of genetic and environmental factors to influence asthma endotypes or biological processes [
14,
15]. The main types of the latest classification of allergic respiratory diseases focus on the characterization of the endotype based on distinct functional or pathophysiological mechanisms and the phenotype based on the description of the condition [
15,
16]. Asthma is characterized by symptoms such as wheezing, chest tightness, shortness of breath, and cough. These symptoms arise due to chronic airway inflammation in response to triggers, commonly inhaled allergens, which lead to airway hyperresponsiveness (AHR), causing variable airway narrowing, airway remodeling, and mucus hypersecretion. The complexity of variation in the symptoms and its pathogenesis make asthma a more complicated heterogeneous syndrome with some endotypes [
17]. The development of chronic airway inflammation due to inappropriate airway immune responses to specific pathogens or allergens has been identified as a crucial pathological feature of asthmatics [
18]. Classical Th2 cell-mediated eosinophilic inflammation is associated with a subset of patients with severe asthma and is primarily identified by IgE levels and blood eosinophil counts in clinical evaluations [
15,
19]. Th2 and non-Th2 are the main endotypes of respiratory allergies. Recently, mixed endotypes such as Th2/Th17 or Th1/Th17 have been proposed [
16]. Th2 cells orchestrate allergic inflammation by releasing Th2 cytokines IL-4, IL-5, and IL-13 [
20,
21]. Th2 cytokines are responsible for eosinophil accumulation and activation, IgE production, and airway abnormalities, including mucus metaplasia, bronchial hyperresponsiveness, and airway wall thickening [
22]. Our study employed the two cell types—lung epithelial cells and lung macrophages—ultimately responsible for the airway inflammation in asthma pathogenesis.
Alveolar epithelial cells (AECs) are composed of type Ⅰ and type Ⅱ cells [
23]. Type Ⅰ AECs form a barrier that is able to sense microbial products, generate inflammatory responses, and participate in gas exchange. Type Ⅱ AECs protect the lungs by secreting antimicrobial molecules such as lysozyme, complement, and surfactant proteins [
24,
25]. Type Ⅱ AECs release various cytokines and chemokines involved in the migration, activation, and differentiation of immune cells. Murine and human type Ⅱ AECs express major histocompatibility complex (MHC) class Ⅱ molecules and can present antigens to CD4+ T cells [
26]. Evidence suggests that asthma is primarily an epithelial disorder. An altered epithelial barrier function exposes the airways, which are fragile in genetically susceptible individuals, to early life virus infections, which stimulate immature dendritic cells and induce local allergen sensitization. If the repair responses of the epithelial barrier are inadequate and there is sustained epithelial susceptibility to allergens, they lead to the persistence of asthma and produce various mediators, leading to airway wall remodeling and prolonged inflammation, which damage the barrier [
27]. Maintaining the structural and functional homeostasis of airway epithelial cells is just as crucial as suppressing excessive inflammation. Once the defense of mucociliary clearance is breached, the airway epithelial cells contact pathogens that infect the respiratory tract [
14,
17]. The airway epithelial cells rapidly respond to microbes, cellular stress, or tissue damage by recognizing pathogen-associated molecular patterns (PAMPs) through membrane-bound or cytoplasmic pattern recognition receptors (PRRs) and initiate appropriate signaling before immune cells are recruited to the infection site. Epithelial PRR activation induces the production of chemokines and cytokines, playing an essential role in initiating allergic responses, which lead to the release of chronic and high levels of pro-inflammatory mediators that can mediate the lung pathology seen in asthma [
18,
27,
28].
Lung macrophages are the most abundant innate immune cells and participate in respiratory defense, displaying a high phagocytic capacity and antigen presentation [
25,
29]. In particular, alveolar macrophages (AMs) participate in pro- and anti-inflammatory functions in allergic asthma [
30]. In both asthmatics and murine models of allergic asthma, after allergen challenge, the rapid recruitment of monocytes occurred and there was an increase in the monocyte-derived population of AMs that promoted acute allergic lung inflammation [
31]. After repeated exposure to allergens, inflammation becomes chronic due to the expanded recruitment of immune cells and consequently elevated levels of cytokines, leading to macrophage polarization [
22]. Significantly, plastic macrophages integrate signals from their microenvironment, leading to context-dependent polarization into M1/classically orM2/alternatively activated macrophages, which represent the two extremes of a broad spectrum of divergent phenotypes [
32]. The polarization is defined mainly by in vitro studies, and tissue macrophages are possibly activated along a continuum between M1 and M2 by variously combined stimuli in vivo [
33]. Therefore, M1 and M2 constitute an inadequate approach for describing the scope of macrophage populations in vivo [
22]. The regulating process of macrophage polarization is a complex interaction among various signaling molecules, including cytokines and chemokines [
33]. The M1 phenotype can be induced by potent stimulants, such as lipopolysaccharide (LPS) and IFN-γ. The M2a subtype of M2 phenotype could be induced by IL-4 or IL-13 [
34,
35]. In general, M1 macrophages are associated with neutrophilic inflammation, and they secrete a quantity of CCL2 and CCL5 chemokines and Th1 cytokines [
33]. On the other hand, M2a cells secrete a quantity of IL-13, CCL17, CCL18, CCL22, and CCL24, which activate Th2 cells and eosinophil inflammation in the lungs [
22,
33]. Unbalanced activation of macrophages is associated with several diseases. In asthma, one or both of these phenotypes are constantly activated, causing chronic inflammation and further damage to the airways [
22,
32]. Previous studies reported distinct macrophage phenotypes in allergic (house dust mite (HDM)-induced) and nonallergic (farm dust extract (FDE)-induced) lung inflammation of murine models. M1 macrophages were more prevalent in the FDE-induced nonallergic model and induced Th1/Th17 lung inflammation associated with neutrophil infiltration and severe asthma with poor corticosteroid response. Meanwhile, M2 macrophages predominated in the lungs of HDM-induced allergic inflammation, which is usually related to Th2 reactions with high levels of serum IgE, significant numbers of eosinophils, and Th2 cytokines. In both models, an increased number of lung macrophages was a common feature [
36,
37].
Chemokines are one of the earliest reactions in the homeostatic and inflammatory responses. The inflammatory reaction is a defensive mechanism to localize and eliminate harmful substances and components of impaired tissue [
38]. Chemokines, signaling proteins that indicate the direction for cell migration, are a large family of small cytokines [
38,
39]. It is a prominent feature of chemokines to recruit leukocytes, and those recruited cells produce other chemokines that contribute to the next wave of leukocyte homing [
39,
40]. The other feature is the complex interactions between chemokines and their receptors [
38]. According to their behavior characteristics, the four main subfamilies (CXC, CC, CX3C, and C) can also be divided into three groups, inflammatory, homeostatic, and dual-type chemokines [
41,
42,
43,
44]. Inflammatory chemokines, such as CCL2, 3, and 4, are induced in leukocytes and other cells in peripheral tissues in response to various threats to the organism, including microbial pathogens, irritants, or toxic cellular components [
41,
44]. They can also be expressed by pro-inflammatory stimuli, such as IL-1, TNF-α, and LPS [
44]. On the other hand, homeostatic chemokines, such as CCL18, 27, and CXCL14, are constitutively expressed by resting cells, mediating the migration and positions of leukocytes in a steady state. The dual-type chemokines, such as CCL20 and 22 and CXCL9 and 10, cover both functions depending on different organs, tissues, or disease conditions. However, these classifications are not fixed and work fluidly [
41,
44]. Chemokine receptors are divided into two groups based on their mechanisms of action: G protein-coupled receptors (GPCRs) activating signals through G proteins, and atypical chemokine receptors (ACKRs) acting by binding with β-arrestin [
45,
46].
Although chemokines play essential roles in inflammatory responses, few studies have been performed on the anti-inflammatory effects and mechanisms of CP and OA on chemokine gene expression profiling. Functional analysis of the overall profile of chemokines and their receptors is more important than the analysis of specific chemokines. We hypothesized that CP and OA may demonstrate their anti-inflammatory effects by modulating inflammatory chemokines and their receptors in the lung inflammation shown in asthma. Therefore, in this study, we aimed to assess the anti-inflammatory effects and mechanisms of CP ethanol extract and OA on chemokines and their receptors’ gene expression utilizing LPS-stimulated mouse lung epithelial cells and lung macrophages in vitro. In particular, we conducted comprehensive gene expression profiling on the chemokines and their receptors in mouse lung epithelial cells. We tried to characterize the classical Th2 and non-Th2 endotypes with chemokines discriminately from the classification using interleukins.
2. Materials and Methods
2.1. Chemicals and Reagents
Oleic acid (Cat. no. O1383), lipopolysaccharides (LPS, Cat. no. L5418), and dimethyl sulfoxide (DMSO, Cat. no. D2650) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). RPMI 1640 Medium with HEPES, RPMI 1640 Medium, antibiotic-antimycotic (100×), and SYBR Green PCR MasterMix were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from Welgene (Gyeongsan-si, Korea). An RNeasy® Mini Kit (Cat. no. 74104), RT2 First Strand kit (Cat. no. 330401), RT2 Profiler PCR Arrays (Cat. no. RAMM-022ZC), and RT2 SYBR Green/RoxqPCRMastermix (Cat. no. 330524) were purchased from Qiagen Sciences Inc. (Germantown, MD, USA). An EZ-Cytox cell viability assay kit was purchased from Daeilab Service co., Ltd. (Seoul, Korea). Ambion TRIzol® reagent was purchased from life technologies (Grand Island, NY, USA). ReverTraAce cDNA synthesis kit was purchased from Toyobo Co., Ltd. (Osaka, Japan). The other chemicals were all analytical or cell culture grade and were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Primary antibodies against iNOS, Cox2, Phospho-Erk1/2 (Thr202/Tyr204), Erk1/2, Phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK, Phospho-SAPK/JNK (Thr183/Tyr185), SAPK/JNK, Phospho-NF-κB p65 (Ser536), NF-κB p65, Phospho-Akt (Ser473), Akt, Phospho-PI3K p85 (Tyr458)/p55 (Tyr199), PI3K p85, PI3K p55, β-actin, Vinculin, Cofilin, GAPDH, C23, and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). ELISA kits were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). Oligonucleotide primers were synthesized from Genotech Corporation (Daejeon, Korea).
2.2. Test Sample Preparation
Cicadidae Periostracum (CP) was purchased from Jeil Pharmaceutical Co. (Wonju, Korea). The dried and chopped CP was extracted 3 times with 70% ethanol using a 3 h reflux. The materials were filtered under reduced pressure in a vacuum rotary evaporator (BUCHI B-480, Buchi, Flawil, Switzerland) at 40 °C. Then, they were dried to yield the CP extract in a freeze-dryer (EYELA FDU-540, Tokyo, Japan), and the extract was stored at 4 °C. The yield of the extract from the initial crude material was roughly 5%. The CP extract and OA were each dissolved in DMSO for the experiment, and the final concentration of DMSO did not exceed 0.2%. All stocks were stored at –20 °C.
2.3. Cell Culture and Sample Treatment
We obtained the mouse lung epithelial cell line (LA-4) and mouse alveolar macrophage cell line (MH-S) from the Korean Cell Line Bank (Seoul, Korea). LA-4 cells were maintained in complete RPMI-1640 with HEPES supplemented with 10% FBS, 100 units/mL of penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL of amphotericin B in a humidified incubator (37 °C and 5% CO2). MH-S cells were maintained in complete RPMI-1640 supplemented with 10% FBS, 0.05 mM of β-mercaptoethanol, 100 units/mL of penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL of amphotericin B in a humidified incubator at 37 °C and 5% CO2. Before treatment, cells were plated at 5 × 105 cells/mL density and incubated overnight. Both cell lines were stimulated with LPS at a concentration of 0.5 μg/mL for 1 h, followed by CP or OA treatments at the indicated concentrations, and incubated at 37 °C in a 5% CO2 incubator for 6 h or 24 h for RNA samples or protein samples, respectively.
2.4. Water Soluble Tetrazolium Salt (WST) Cell Viability
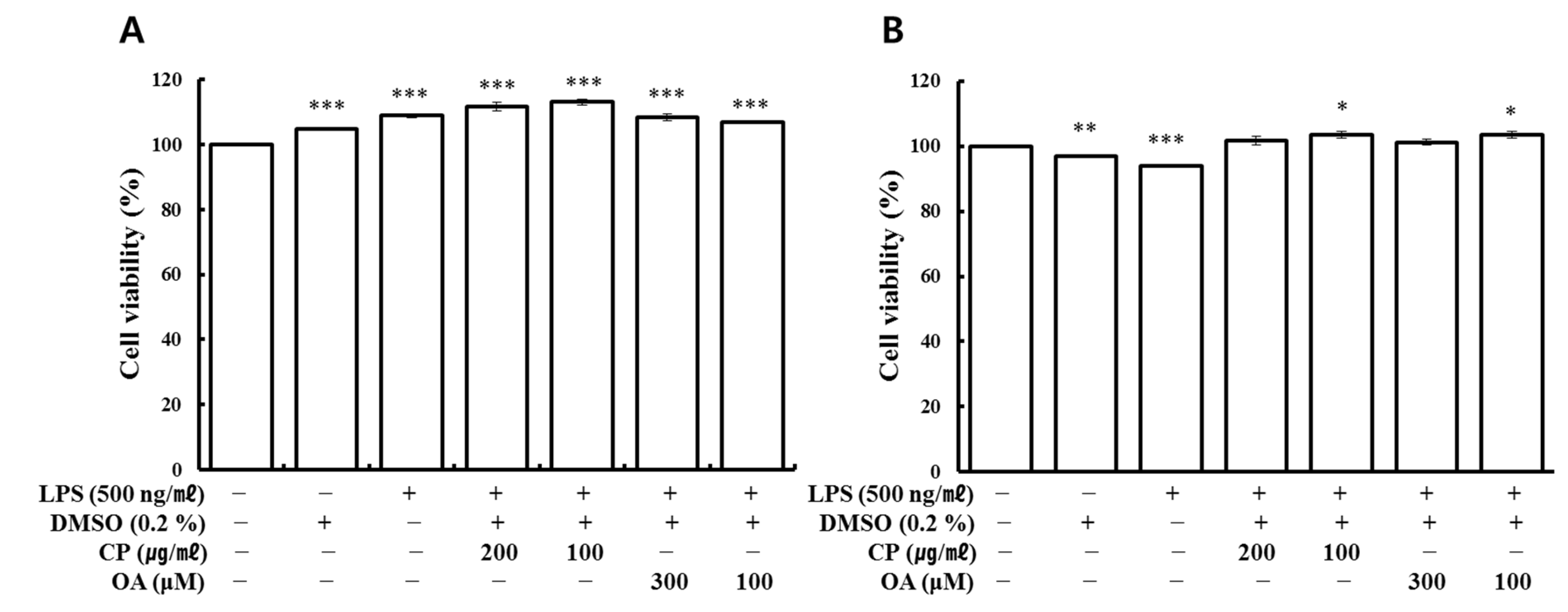
Cell viability was analyzed using the water-soluble tetrazolium (WST) assay. Cells were seeded at a density of 1 × 104 cells/well in 96-well culture plates overnight. Then, they were exposed to various concentrations of the test compounds with or without LPS. Cells were incubated for 24 h at 37 °C in a 5% CO2 incubator, and during the last 4 h, WST (Ez-cytox) solution was added to each well according to the manufacturer’s instructions. Then, absorbance was read by a microplate reader (Epoch micro-volume spectrophotometer system, BioTek Inc., Santa Clara, CA, USA) at 450 nm and 600 nm as a reference. The background was adjusted by measuring the absorbance of cell-free medium containing WST solution.
2.5. RNA Extraction and Gene Expression Profiling Analysis
Total RNA was isolated using the RNeasy® Mini kit. Then, the RNA was reverse transcribed into template cDNA from 0.5 μg total RNA of each sample for a 96-well plate (an array) using the RT2 first strand kit according to the manufacturer’s instructions. Briefly, cells were first lysed in buffer RLT, and the lysate was mixed with ethanol and then centrifuged using an RNeasy Mini spin column to bind the RNA to the silica membrane in the column. Traces of DNA that might co-purify with the RNA were removed by DNase treatment on the silica membrane. DNase and any contaminants were efficiently washed away, and high-quality total RNA was eluted in RNase-free water. Total RNA was quantified using a microplate reader (Epoch micro-volume spectrophotometer system, BioTek Inc., Santa Clara, CA, USA). Finally, the transcribed cDNA was ready for use in the downstream application according to the following two steps: elimination of genomic cDNA contamination and reverse transcription. The template cDNAs were characterized in technical triplicate using the Mouse Chemokine and Receptors RT2 Profiler PCR Arrays with RT2 SYBR Green/Rox qPCR Mastermix on the StepOnePlus Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The RT2 Profiler PCR Array plate contains the predispensed gene-specific primer sets for 84 genes that encode mouse chemokines and their receptors, as well as other related genes. In addition, each array includes five housekeeping genes and three RNA quality control elements for monitoring: genomic DNA contamination control (GDC), reverse transcription controls (RTC), and real-time PCR efficiency control (PPC). According to the manufacturer’s protocol, RNA quality was determined using RT2 RNA QC PCR arrays. The following PCR conditions were used: hold for 10 min at 95 °C and 60 s at 60 °C. The threshold was manually defined using the log view of the amplification plots, and the threshold cycle (CT) cut-off was set to 35. qPCR was performed in triplicate for each gene, and the quality was controlled by verifying a single peak in the melting curve analysis.
The CT values of target genes were normalized to an average of two housekeeping genes, GAPDH and ACTB. The comparative CT method was then used to examine the relative quantification of the samples. Fold changes in gene expression were then calculated for pairwise comparison using the equation 2-ΔΔCT. Fold change values greater than 1 indicate a positive or up-regulation, and fold change values less than 1 indicate a negative or down-regulation. The fold regulation is the negative inverse of the fold change. Fold regulation represents fold-change results in a biologically meaningful way. For example, a fold regulation value of −4.0 occurred when the normalized expression of a target gene in a test group was 4 times lower than that in the control group. The fold regulation threshold cut-off was set to at least 1.5.
2.6. Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
The total RNA of LA4 and MH-S were extracted using Ambion TRIzol
® reagent and reverse-transcribed into cDNA using a ReverTraAce cDNA synthesis kit according to the manufacturer’s protocol. Total RNA was quantified using a microplate reader (Epoch micro-volume spectrophotometer system, BioTek Inc., Santa Clara, CA, USA). A total of 100 ng of cDNA and 10 pmole of gene-specific forward and reverse primers were loaded in the qPCR system (StepOnePlus Real-Time PCR system, Applied Biosystems, Foster City, CA, USA). Different types of chemokines and cytokines were amplified with SYBR Green PCR MasterMix. Mouse primer sequences for qRT-PCR are listed in
Table 1. The oligonucleotides were manufactured by Genotech Corporation (Daejeon, Korea). The following amplification protocol was used: 2 min at 50 °C, 2 min at 95 °C, then 40 cycles of 15 sec at 95 °C and 1 min at 60 °C. The CT values of target genes were normalized to that of GAPDH. For each gene, qPCR was performed in triplicate, and the quality was controlled by verifying a single peak in the melting curve analysis. The comparative CT method was then used to examine the relative quantification of the samples.
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of different chemokines and cytokines in cell culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions using antibody pairs from R&D Systems, Inc. (Minneapolis, MN, USA). Cell culture supernatants were collected, and the debris was removed by centrifugation at 1000× g for 10 min at 4 °C, followed by –80 °C freezing before analysis. The optical density of each well was immediately determined using a microplate reader (Epoch micro-volume spectrophotometer system, BioTek Inc., Santa Clara, CA, USA) at the recommended wavelength of the manufacturer.
2.8. Western Blot Analysis
Cells were lysed with RIPA lysis buffer (Sigma Chemical Co., St. Louis, MO, USA) for whole lysates, and the cell debris was removed via micro-centrifugation at 8000× g for 10 min at 4 °C. For cell fractionation, cytoplasmic and nuclear extracts were separated using the Nuclear/Cytosol Fractionation Kit (Biovision Incorporated, Milpitas, CA, USA) according to the manufacturer’s instructions. The protein concentration was determined by the Bradford method using Bio-Rad protein assay reagent (Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to the manufacturer’s protocol. Total protein was diluted with sample buffer stock solutions (Laemmli’s SDS-sample buffer) to a final sample buffer concentration of 1× and incubated at 100 °C for 10 min. The prepared protein samples were subjected to various quantities (4–20%) of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel depending on the size of the protein of interest. Electrophoresed proteins were transferred onto nitrocellulose membranes using an electrophoretic transfer system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Then, the membranes were blocked with 5% skim milk in Tris-buffered saline (50 mM Tris-HCL, 150 mM NaCl, pH 7.5) with 0.1% Tween 20 for 1 h at room temperature. The membranes were then incubated overnight with each specific primary antibody at 4 °C. After washing, the membranes were incubated with each specific horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature. Finally, the blots were developed via enhanced chemiluminescence detection reagents (GenDEPOT, Katy, TX, USA). The densities of the bands on immunoblots were measured by Image J (available from the National Institutes of Health).
2.9. Statistical Analysis
For the RT2 Profiler PCR Array, p-values were calculated based on a Student’s t-test of triplicate 2^ (-Delta CT) values for each gene between 2 comparison groups, and only p-values less than 0.05 were included in the data. The data are presented as the mean ± SEM (standard error of the mean) of triplicate experiments for the other experiments. Statistically, significant differences were assessed by one-way analysis of variance (ANOVA), followed by Student’s t-test using SPSS statistical analysis software (version 14.0, IBM, Armonk, NY, USA). Statistical significance is indicated in the figures and tables.
4. Discussion
Chemokines convey information to leukocytes in the homeostatic or inflammatory environment and control the immune responses through chemokine receptors widely expressed on immune cells. Chemokines and chemokine receptors are intensively involved in innate and adaptive immune responses [
41]. In this study, we aimed to assess the anti-inflammatory effects and mechanisms of CP and OA treatments in LPS-induced lung and airway inflammation in vitro. In particular, we conducted comprehensive gene profiling on the chemokines and their receptors in mouse lung epithelial cells. Then, we tried to characterize the classical Th2 and non-Th2 endotypes with chemokines discriminately from the classification using interleukins.
LPS is a highly immunogenic antigen with the ability to stimulate the host cells of the innate immune system via toll-like receptor (TLR) 4, which is one of the PRRs implicated in inflammatory pathways. TLR4 recognizes common PAMPs presented on phagocytic and epithelial cells [
100,
101]. This stimulation results in various pro-inflammatory cytokines and chemokines that mediate inflammation and is strongly related to lung damage [
71,
102]. Therefore, LPS stimulation of lung epithelial cells and lung macrophages in vitro can mimic lung inflammation. Lung epithelial cells are constantly exposed to microbial challenges due to respiration and are responsive to the TLR4 activator LPS [
102,
103]. LPS induced neutrophil accumulation and increased the expression of pro-inflammatory molecules in rodents [
104], possibly by activating TLR4/NF-κB signaling pathways, leading to inflammation [
104,
105].
The response to LPS is dependent on binding to the membrane receptor CD14 in the presence of LPS binding protein (LBP) in serum. CD14, along with TLR4 and myeloid differentiation factor 2 (MD-2), functions as a co-receptor for LPS detection for signal transduction. TLR4, with CD14, plays a role in the cellular response to pathogens as the primary mediator of LPS signaling, followed by transcription factor NF-κB activation and cytokine production [
106,
107].
Interestingly, LPS stimulation significantly downregulated CXCL12 expression in our study (
Table 3). CXCL12 acts via CXCR4 and ACKR3 receptors, which are significantly expressed on the cancer cell surface [
108]. Classically, CXCL12 recruits stromal cells in the lungs under normal conditions, and CXCR4 has been implicated in more than 20 human cancers. As the CXCL12/CXCR4 axis correlates with the angiogenesis, proliferation, and metastasis of tumors, the axis is attracting increasing attention as a potential therapeutic target [
109]. However, there is some evidence of the opposing functions of CXCL12, so it is somewhat controversial. Some suggested that a reason for this discrepancy may be due to the existence of at least six CXCL12 isoforms in humans and three isoforms in mice, each with a different role [
45,
108]. In our study, the decreased expression of CXCL12 in the LPS-stimulated group may be explained by the following two studies.
The response to LPS by the innate immune system is mainly dependent on TLR4, along with CD14 and MD-2. Additional receptors, such as CD55, CXCR4, and heat shock proteins (HSP), have been proposed as part of the activation cluster. Triantafilou et al. suggested that CXCR4 seems to be crucial for LPS signaling with other LPS co-receptors [
110]. In addition, a previous study demonstrated that the expression levels of inflammatory factors such as TNF-α, IL-6, IL-8, COX-2, and NF-κB were downregulated by a CXCR4 antagonist drug [
111]. Dai et al. emphasized the CXCL12/CXCR4 autocrine loop, which significantly promotes the motility, proliferation, and invasiveness of non-small cell lung cancer (NSCLC). The findings suggest that therapies related to CXCR4 in the treatment of NSCLC should consider CXCL12’s expression status. This is because autocrine overexpression of CXCL12 in some tumors may be more sensitive to CXCR4 antagonists by competing with CXCL12 for receptor binding, meaning that they may be more suitable for the application of chemokine-based anti-cancer therapies [
112]. Therefore, in our study, LPS probably interfered with the autocrine expression of CXCL12 in the CXCL12/CXCR4 axis via competition for CXCR4 binding. Thus, LPS may downregulate CXCL12 expression, whereas CXCR4 may act as one of the co-receptors for LPS binding. Consequently, LPS may act as an antagonist to CXCR4. Scrutiny of those mechanisms would be an excellent topic for further study.
In the other study, the expression of CXCL12-γ in the central nervous system (CNS) was significantly decreased in experimental autoimmune encephalomyelitis (EAE)-prone Dark Agouti (DA) rats compared to that of EAE-resistant Albino Oxford (AO) rats. In CNS, inhibition of nitric oxide (NO) synthesis in DA rats upregulated CXCL12-γ expression, while the contribution of NO in AO rats downregulated CXCL12-γ expression. Moreover, NO remarkably inhibited CXCL12-γ expression in astrocytes in vitro, suggesting its modulatory effect on CXCL12-γ expression in neuroinflammation [
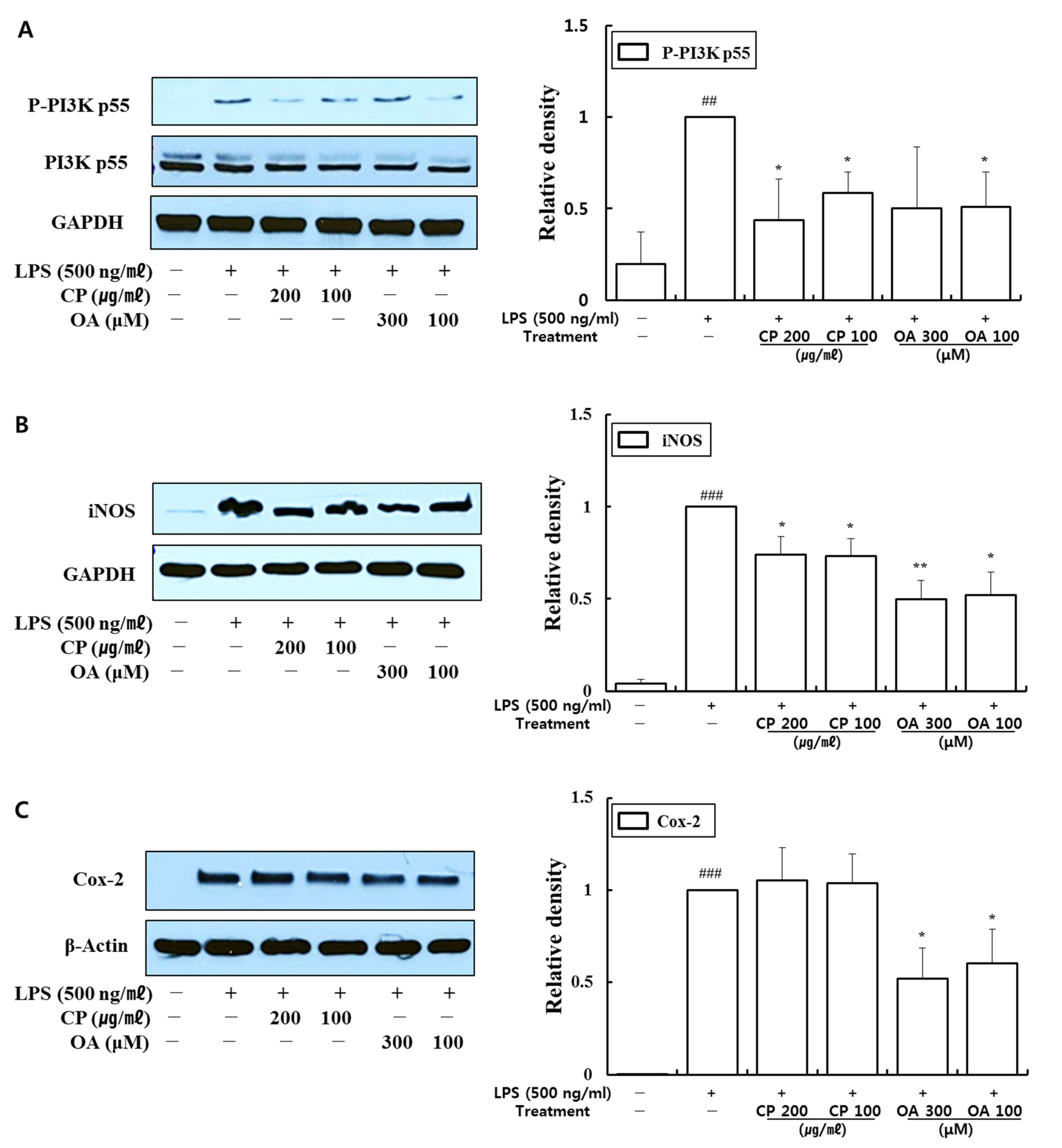
113]. It can be assumed that a similar mechanism would have worked in our study as well since the expression of iNOS in LPS-stimulated macrophage cells was significantly increased compared with that in the normal group in the Western blot results (
Figure 9). A similar pattern was also confirmed in our preliminary data for the lung epithelial cells (data not shown).
Asthma is also a multicellular disease because it involves integrative reactions to triggers in the airways, which cause abnormal or excessive responses by various cell types, including mast cells, eosinophils, dendritic cells, neutrophils, macrophages, T lymphocytes, and other airway constituent cells such as airway smooth muscle cells and airway epithelial cells [
20,
114]. The highest numbers of eosinophils and T lymphocytes were found in bronchial biopsies of allergic and nonallergic asthma [
115]. CD4+ T cells can differentiate into one of several distinct T cell types functionally when they come across antigens. In particular, Th1 cells typically express CCR1, CCR5, CXCR3, and CXCR6; Th2 cells preferentially express CCR3, CCR4, and CCR8; and Th17 cells express CCR6. These effector T cells can migrate to inflamed tissues via their receptors, contributing to microbial clearance and tissue repair. In particular, CXCL9, CXCL10, and CXCL11 are associated with Th1 cell functions; CCL1, CCL17, CCL18, and CCL22 are associated with Th2 cell functions; and CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL8 are associated with Th17 cell functions. In addition, CCL4 was significantly associated with a mix of lymphocytes, including Th1, Th2, Th9, and Th17, in severe asthma patients [
16,
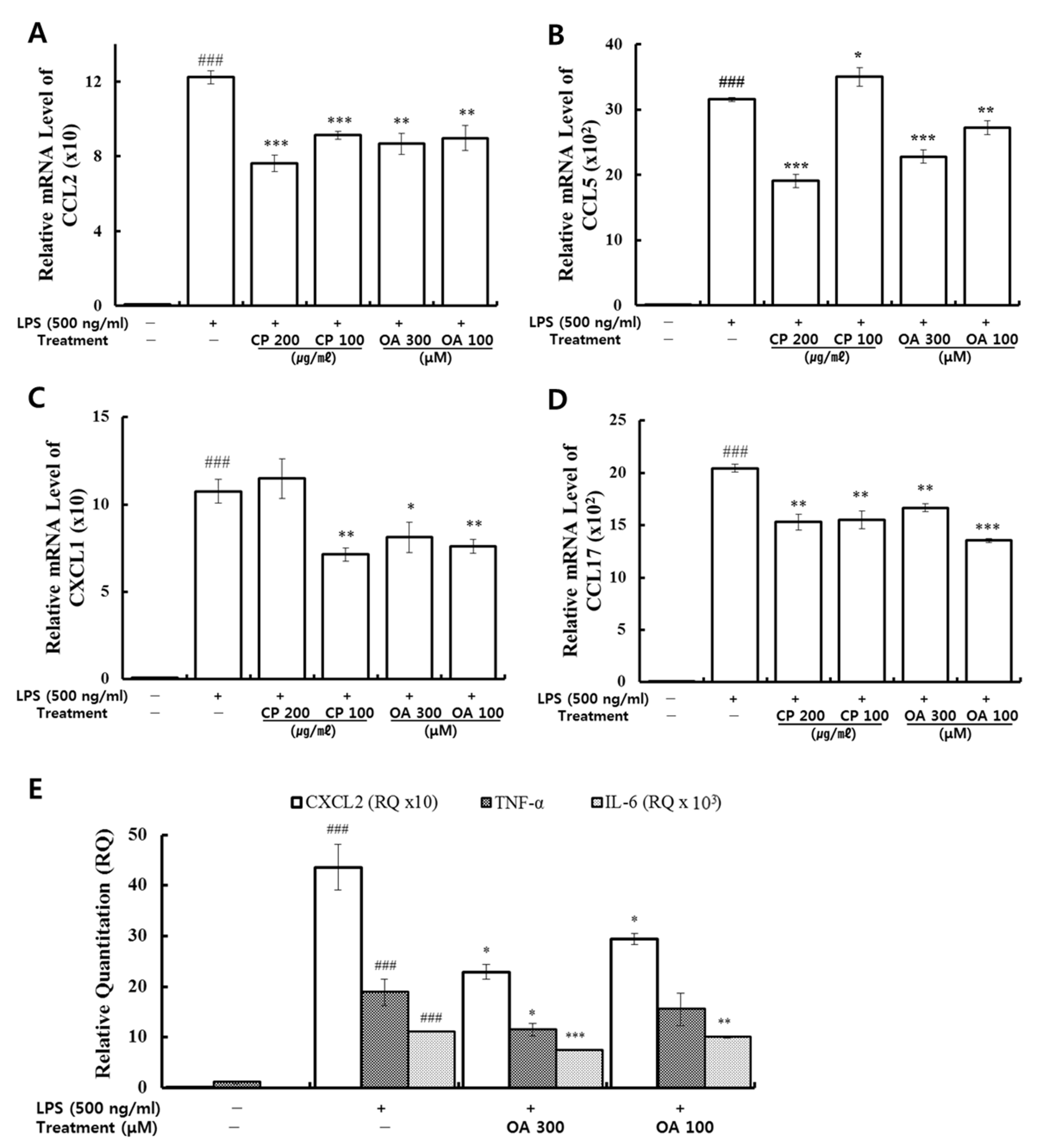
39]. Furthermore, expression levels of CCR3 are high in eosinophils. Eosinophil migration is mediated by a gradient of CCL3, CCL7, and CCL22, mainly produced by interstitial macrophages, and the gradient of these chemokines can be changed by the chemokines CCL5 and CCL11, which are released at high levels by the airway epithelium [
55]. Despite the application of standard therapy with bronchodilators and inhaled corticosteroids, approximately 10% of asthmatics have persistent severe symptoms [
20]. Given the vital roles of chemokines in the immune system and during inflammatory responses, a number of chemokines and GPCRs seem to have an essential contribution to the pathogenesis of asthma [
116]. These molecules have become the most prominent targets for drug development because of their active participation in disease progression [
109]. At present, many chemokine inhibitors are under development for the treatment of asthma. For example, anti-CCR3 (ASM8) and anti-CCL11 (Bertilimumab) are in phase 2 clinical trials [
57].
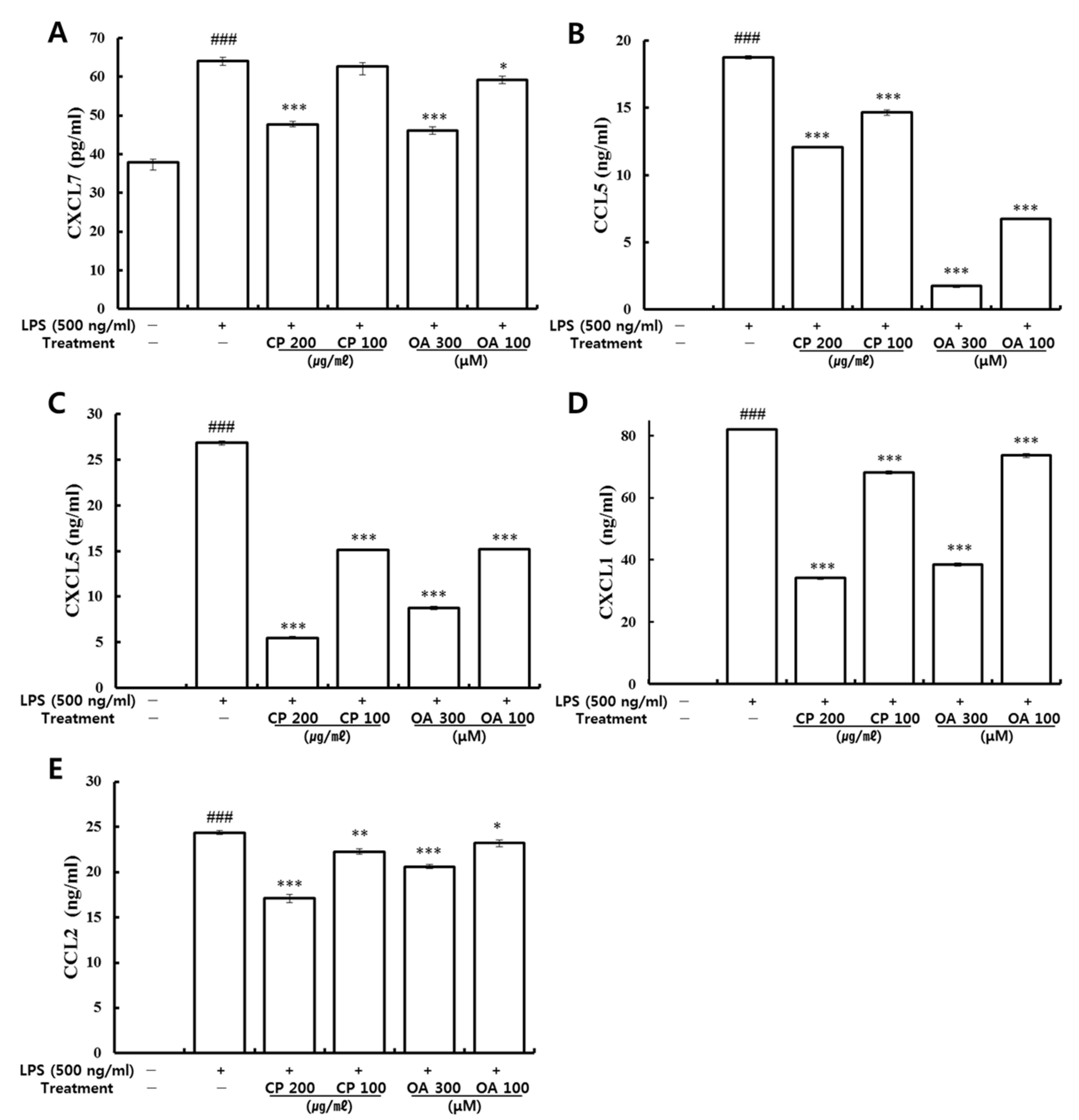
Through LPS stimulation, LA4 cells (type Ⅱ-like AECs) express diverse chemokines, which can chemoattract various innate and adaptive immune cells [
26,
71,
117,
118]. The inhibition of inflammatory chemokines apparently affects the local inflammatory response by interfering with immune cell recruitment. In allergic asthma, a significant negative correlation was found between the numbers of T lymphocytes and eosinophils and epithelial integrity [
115].
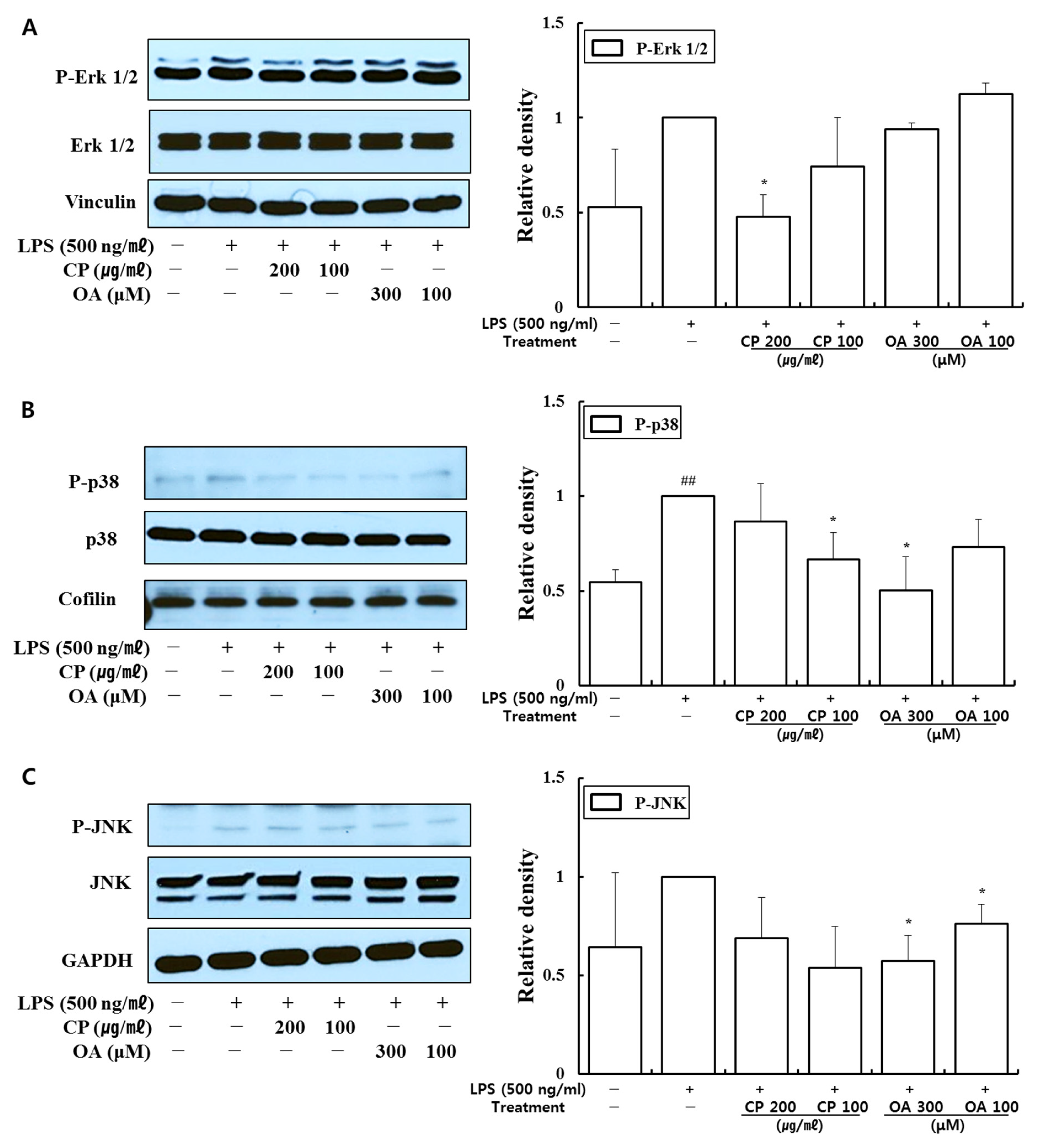
Targeted therapies that modulate cell signaling pathways can be a powerful strategy to treat asthma. Thus, signaling molecules can be potential targets for treating asthma. MAPK signaling pathways regulate immune responses and inflammation by controlling the gene expression of inflammatory factors in asthma. Fengjuan et al. reported significant increases in IgE and IL-4 and inflammatory mediators such as IL-6, IL-17, TNF-α, and NO in an OVA-induced rat asthma model. IL-4 and IgE could be regulated by inhibiting the phosphorylation of Erk, JNK, and p38, and the nuclear translocation of phospho-p65 [
119,
120]. The IL-4/IL-13/STAT-6 pathway is a well-known key regulator in asthma pathophysiology. MAPKs transactivate STAT-6 by phosphorylating its serine residues. In particular, the inhibition of STAT-6 phosphorylation by Erk and p38 inhibitors suppressed the expression of IL-4/IL-13-induced inflammatory chemokines such as CCL2, CCL11, CXCL1, and CXCL3. Thus, inhibitors of Erk and p38 can be considered as potential therapeutic agents in asthma [
119]. JNK also plays a meaningful role in the airway remodeling and apoptotic process by inducing the Wnt5a/JNK signaling pathway in asthma [
121]. Furthermore, considerable research efforts have revealed that the phosphorylation of NF-κB contributes to controlling NF-κB-directed transactivation. Crucially, NF-κB phosphorylation provides new opportunities to selectively target NF-κB as therapeutic targets by controlling transcription in a gene-specific manner [
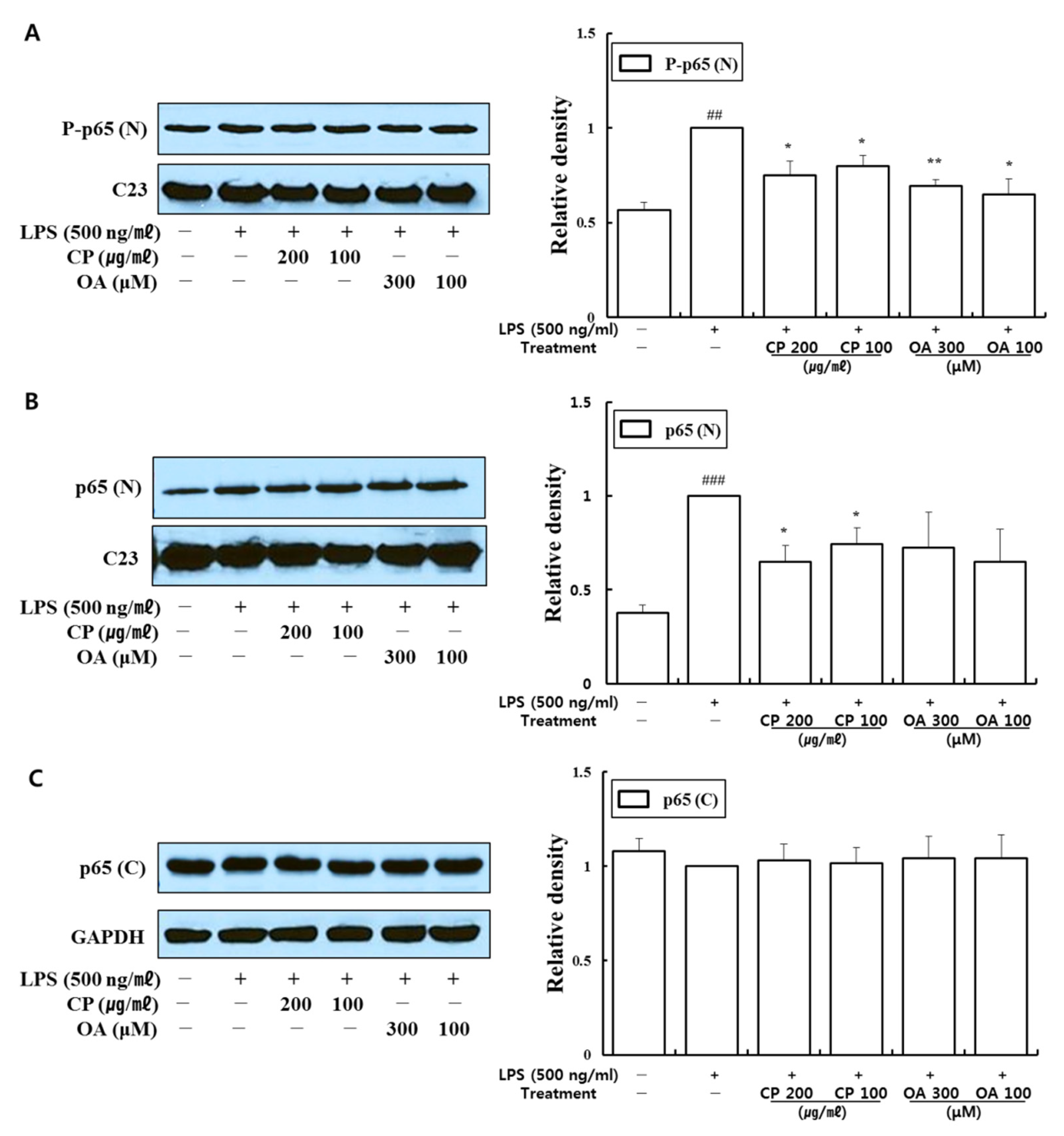
122]. Indeed, previous studies have found that the phosphorylation of p65 at S536 is not regulated by, or associated with, IκBα. Instead, this phosphorylation may lead to the expression of a distinct set of target genes [
122,
123]. In addition, the PI3K/Akt signaling pathway contains upstream molecules of NF-κB, which has a regulatory role in allergic asthma [
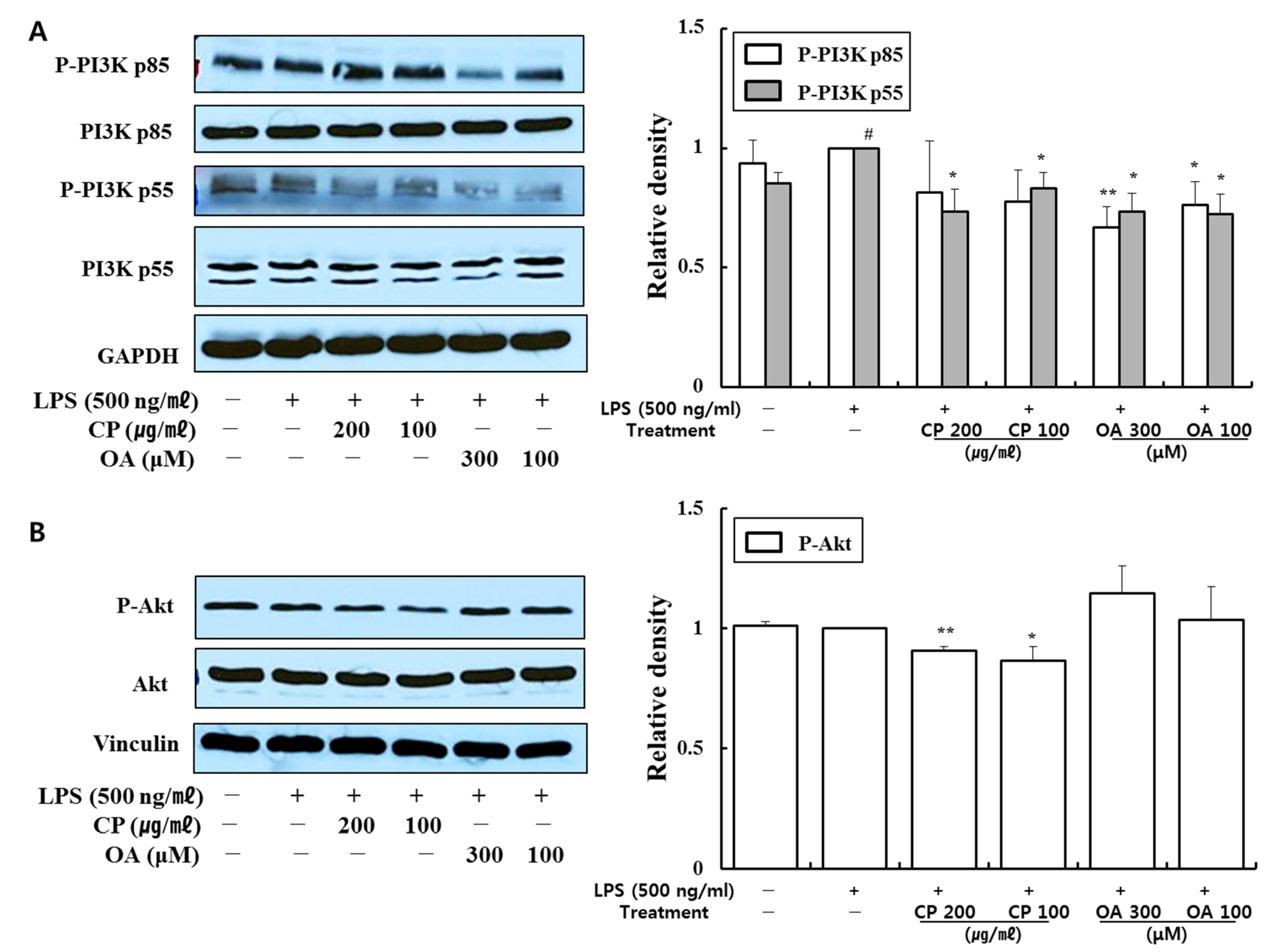
97,
124,
125,
126,
127].
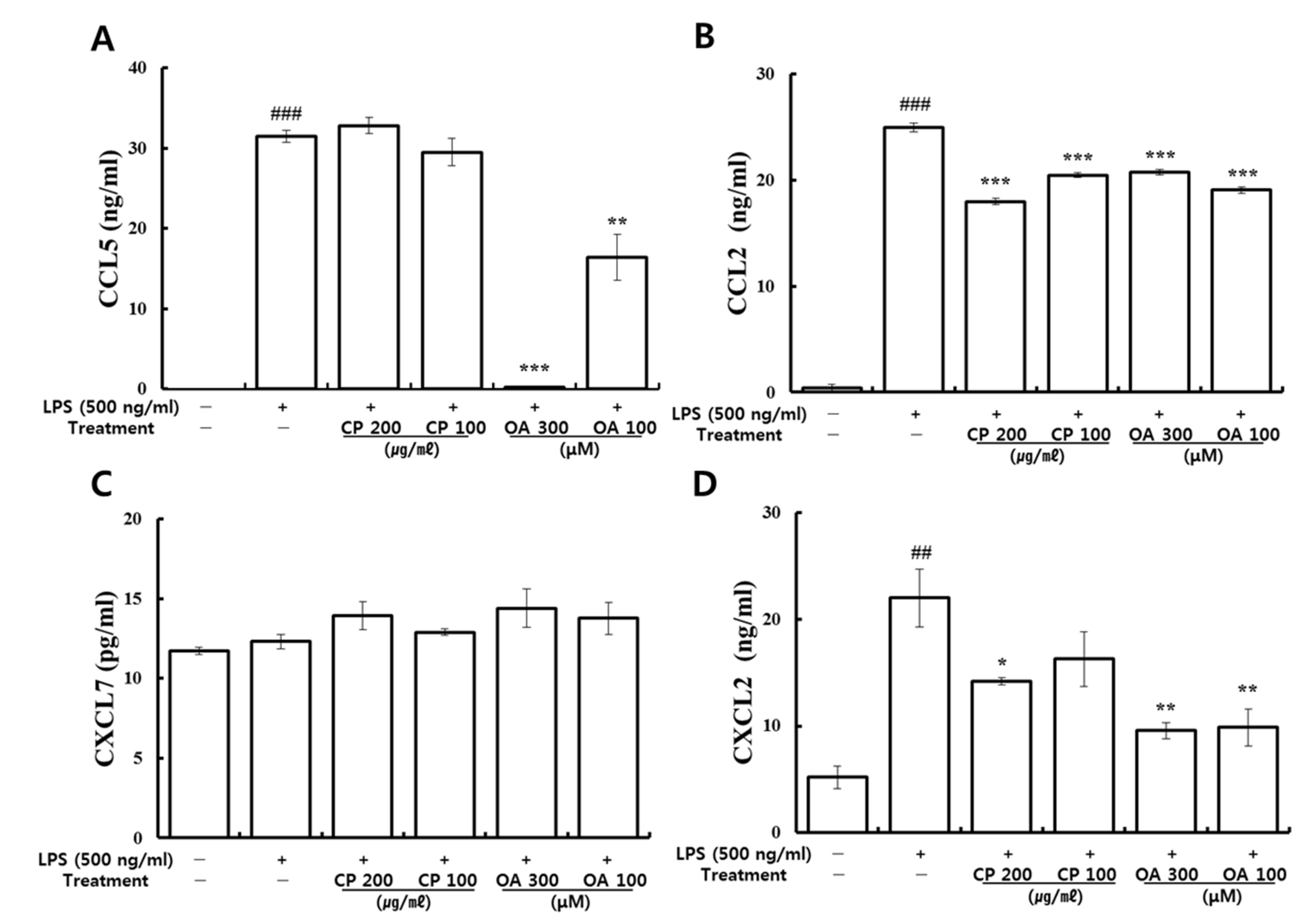
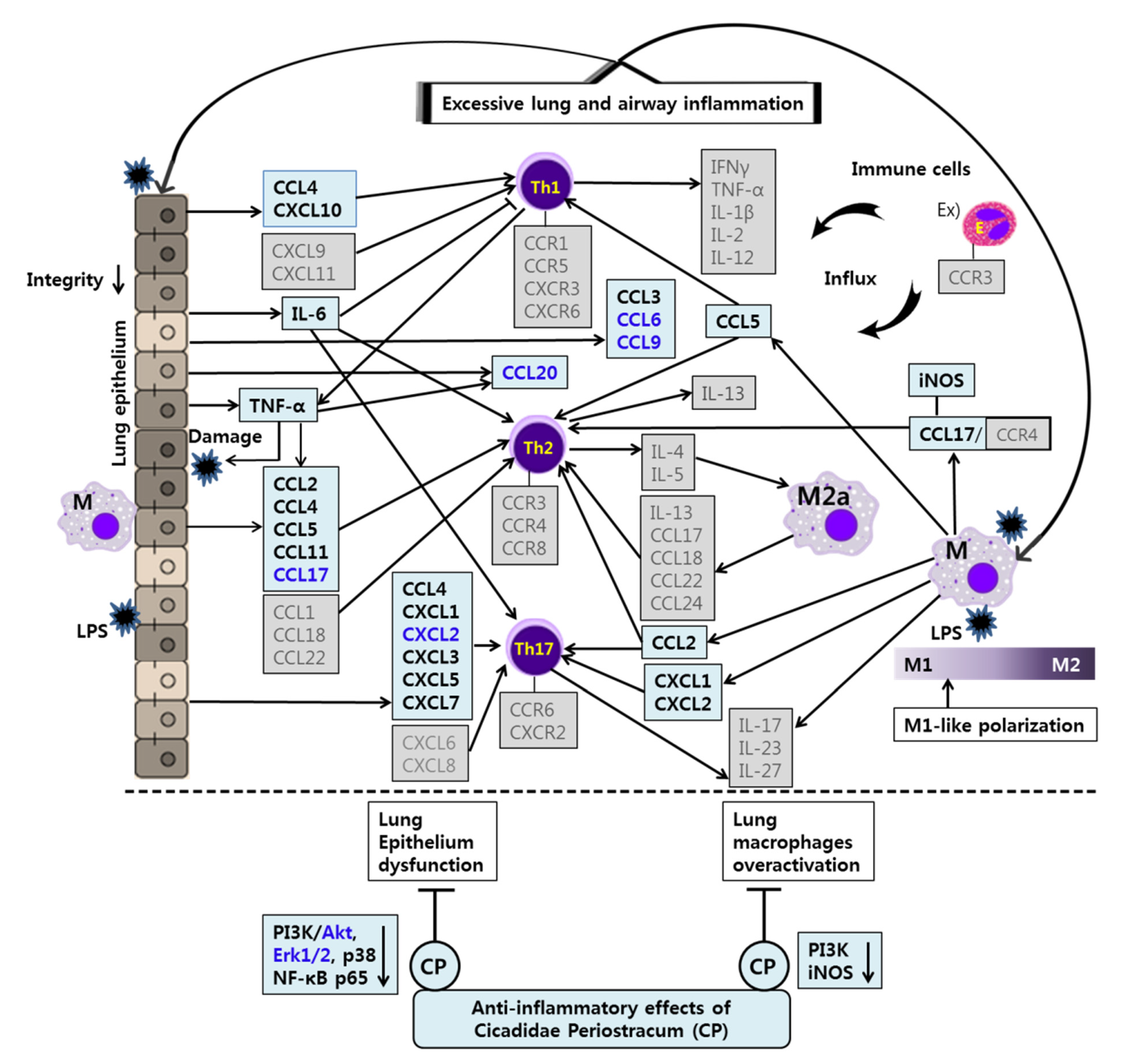
In our study, CP treatments probably exhibit their anti-inflammatory effects by suppressing the recruitment of mast cells, dendritic cells, NK cells, monocytes, macrophages, eosinophils, neutrophils, basophils, and lymphocytes, and by modulating T cell differentiation, dendritic cell maturation, and Th1/Th2/Th17 lymphocytes according to the roles of the reversed chemokines (
Table 4 and
Table 5,
Figure 2 and
Figure 3), as described in the results. These results prove that CP treatments exhibit anti-inflammatory effects on suppressed inflammatory chemokines and cytokines by inhibiting pro-inflammatory mediators, including PI3K-p55, Akt, Erk1/2, p38, and NF-κB, in the LPS-stimulated LA4 cells (
Figure 4,
Figure 5 and
Figure 6).
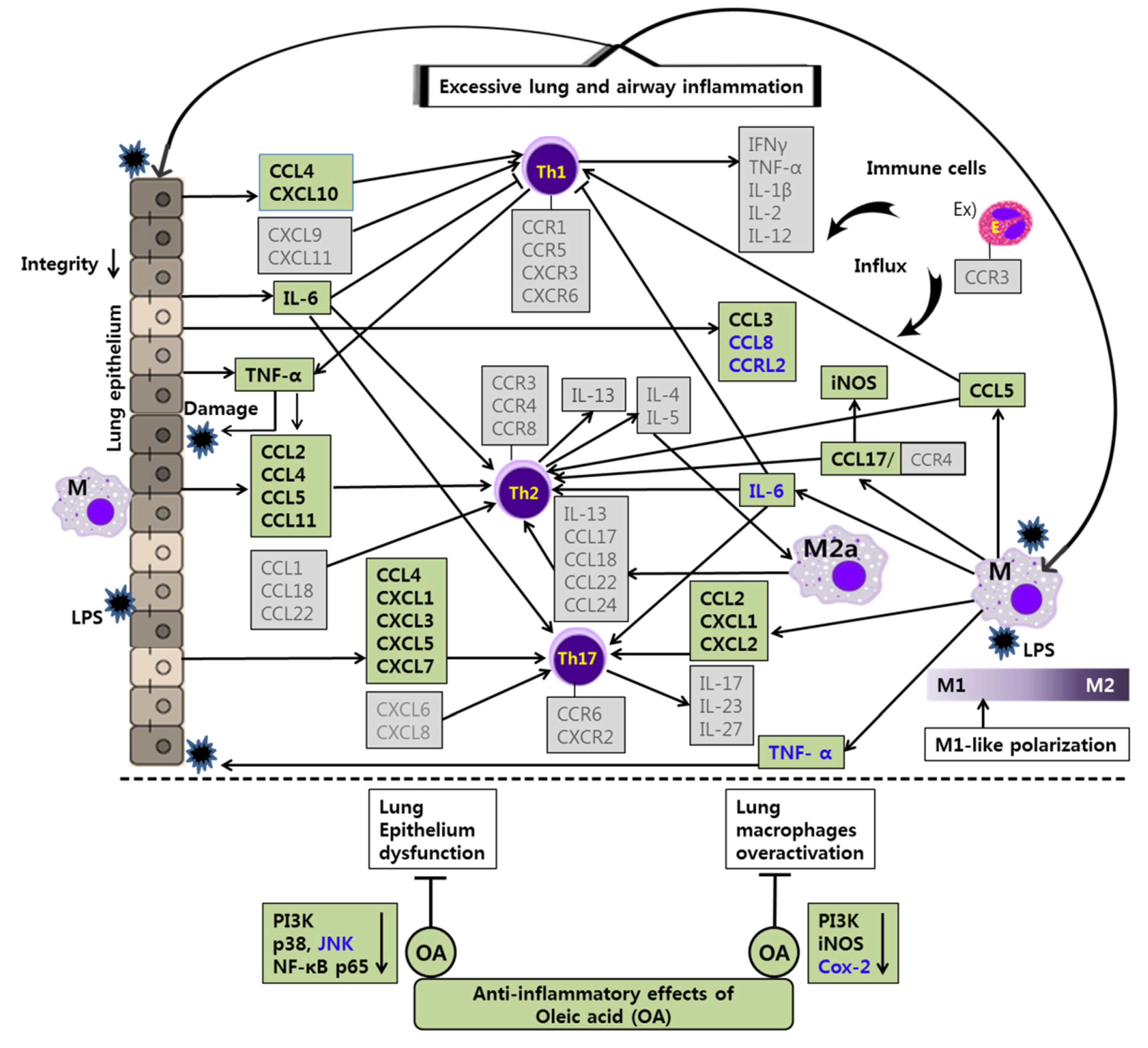
Likewise, OA treatments possibly exhibit anti-inflammatory effects by suppressing the recruitment of mast cells, dendritic cells, NK cells, neutrophils, basophils, eosinophils, monocytes, macrophages, lymphocytes, and by modulating Th1/Th2/Th17 lymphocytes functions according to the roles of the reversed chemokines (
Table 6 and
Table 7,
Figure 2 and
Figure 3), as described in the results. These results prove that OA treatments display anti-inflammatory effects on inflammatory chemokines, cytokines, and a receptor by inhibiting pro-inflammatory mediators, including PI3K-p85, PI3K-p55, p38, JNK, and NF-κB, according to the data (
Figure 4,
Figure 5 and
Figure 6). It is notable on the mechanisms of action of CP and OA concerning the inhibition of PI3K.
Interesting studies have been published regarding the inhibition of CCRL2 by OA treatment (
Table 6). CCRL2 belongs to the group of ACKRs. ACKRs are devoid of chemotactic activity and are characterized by their ability to scavenge chemotactic factors from inflamed tissues. Despite being in the group of ACKRs, CCRL2 does not bind chemokines and is devoid of scavenging functions, but does modulate leukocyte migration. CCRL2 is expressed by monocytes/macrophages, mast cells, dendritic cells, bronchial epithelia, endothelial cells, microglia, and astrocytes in mice. In studies, CCL2, CCL5, CCL7, CCL8, and CCL19 were proposed as its ligands, but this was not subsequently confirmed. Thus far, only chemerin, a non-chemokine chemotactic protein, has been generally accepted as a CCRL2 ligand. CCRL2 remains unexplored in various aspects, but its roles in relation to inflammation, leukocyte migration, and tumors have been reported [
128,
129]. The expression of CCRL2, which has been reported to form a heterodimer with CXCR2, was rapidly upregulated by LPS, TNF, and other inflammatory stimuli in neutrophils and dendritic cells and induced in lung macrophages and the bronchial epithelia in OVA-induced airway inflammation [
130,
131]. Otero et al. demonstrated the unique role of CCRL2 in lung DC migration using CCRL2 KO mice. CCRL2 KO mice showed a marked reduction in leukocyte recruitment in the bronchoalveolar lavage, particularly with regard to mononuclear cells and eosinophils in a Th2 model of OVA-induced airway inflammation, whereas regular recruitment of circulating DCs into the lungs did occur. This reduction in leukocyte recruitment was caused by decreased production of Th2 cytokines (IL-4 and IL-5) and chemokines (CCL11 and CCL17). The decreased local Th2 response was directly correlated with the reduced migration of antigen-loaded lung DCs to mediastinal lymph nodes and the subsequently decreased priming of antigen-specific T cells in the regional lymph nodes. The study showed the specific role of CCRL2 in lung DCs in terms of their migration into mediastinal lymph nodes and their contribution to excessive lung inflammation [
132].
According to traditional Korean medicine, CP exhibits its efficacy in the lungs and liver. Interestingly, OA, one of the main bioactive components in CP, has shown a specific inhibitory effect on CCRL2 expression in LPS-stimulated epithelial cells (
Table 6). OA treatments may interrupt neutrophil recruitment by decreasing CCRL2. It is possible that OA exerts its inhibitory effect on CCRL2 expression in lung DCs and subsequently contributes to the resistance to allergic asthma by decreasing the migration of lung DCs into mediastinal lymph nodes. Additional follow-up studies investigating the OA mechanism concerning CCRL2 in animal models would be intriguing.
Unbalanced polarization or overall activation of macrophages contributes to chronic lung inflammation in asthma, as with Th1/Th2 polarization of T cells. The complex networks of chemokines and their receptors actively participate in macrophage polarization and activation [
32,
33,
133]. Macrophages have been extensively acknowledged as desirable therapeutic targets for inflammatory diseases [
33].
The expressions of iNOS and Cox-2 are upregulated by activated NF-κB, which are crucial mediators in the development of pulmonary inflammation. Meanwhile, the activation of NF-κB by iNOS and Cox-2 can subsequently induce other inflammatory cells and mediators. Therefore, the regulation of iNOS and Cox-2 is required to control inflammation in the airways and lungs [
119].
In our study, CP treatments probably exhibit anti-inflammatory effects by suppressing the recruitment of macrophages, neutrophils, monocytes, and eosinophils and modulating Th1/Th2/Th17 lymphocytes according to the roles of the reversed chemokines (
Figure 7 and
Figure 8), as described in the results. These results prove that CP treatments exhibit anti-inflammatory effects on suppressed inflammatory chemokines (
Figure 7 and
Figure 8) by inhibiting pro-inflammatory mediators, including phospho-PI3K p55, iNOS, and Cox-2, in LPS-stimulated MH-S cells (
Figure 9). Previous studies showed the suppressed expression of iNOS and Cox-2 by a PI3K inhibitor in the bronchial epithelia of asthmatic rats and particulate matter (PM)-exposed bronchial epithelia, respectively [
97,
134]. Our data may indicate that CP treatments exhibited suppressive effects on iNOS expression by downregulating the phosphorylation of PI3K p55. Likewise, OA treatments could exhibit anti-inflammatory effects by inhibiting the recruitment of macrophages, eosinophils, and neutrophils, monocytes, and by modulating Th1/Th2/Th17 lymphocytes according to the roles of the reversed chemokines and cytokines (
Figure 7 and
Figure 8), as described in the results. These results prove that OA treatments demonstrate anti-inflammatory effects on inflammatory chemokines and cytokines by suppressing pro-inflammatory mediators, including phospho-PI3K p55, iNOS, and Cox-2, in LPS-stimulated MH-S cells (
Figure 9). Our data may indicate that OA treatments exhibited suppressive effects on iNOS and Cox-2 expression by downregulating the phosphorylation of PI3K p55 according to previous studies [
97,
134]. Notably, CP and OA treatments inhibited PI3K as a potential upstream regulator of iNOS and Cox-2 expression in LPS-stimulated MH-S cells.
The anti-inflammatory effects and mechanisms of CP and OA treatments in lung epithelial cells and lung macrophages after LPS stimulation are illustrated in
Figure 10 and
Figure 11. Our integrated data offer a powerful approach that enables us to assess the full spectrum of chemokines in LPS-induced lung inflammation in vitro. These data provide new insights into lung and airway inflammation from the aspect of chemokines induced by lung epithelial cells and lung macrophages (
Figure 10 and
Figure 11). Our results show that CP and OA are potential chemokine-based therapeutic substances for treating lung and airway inflammation, and our findings could lay the groundwork for chemokine-based therapies in lung and airway inflammation if replicated in further animal models. We suggest that CP and OA can be utilized to apply chemokine-based anti-asthmatic therapies, given that our group’s previous study showed the efficacies of CP and OA in asthmatic mice [
7].
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}