
Sputum Microbiota in Coal Workers Diagnosed with Pneumoconiosis as Revealed by 16S rRNA Gene Sequencing


 ,
,  ,
,
Abstract
1. Introduction
2. Methods
2.1. Cohort Information
2.2. Sample Collection, Processing and Storage
2.3. DNA Extraction, 16S rRNA Amplification and 16S rRNA Sequencing
2.4. Taxonomy Quantification Using 16S rRNA Gene Sequences and Statistical Methods
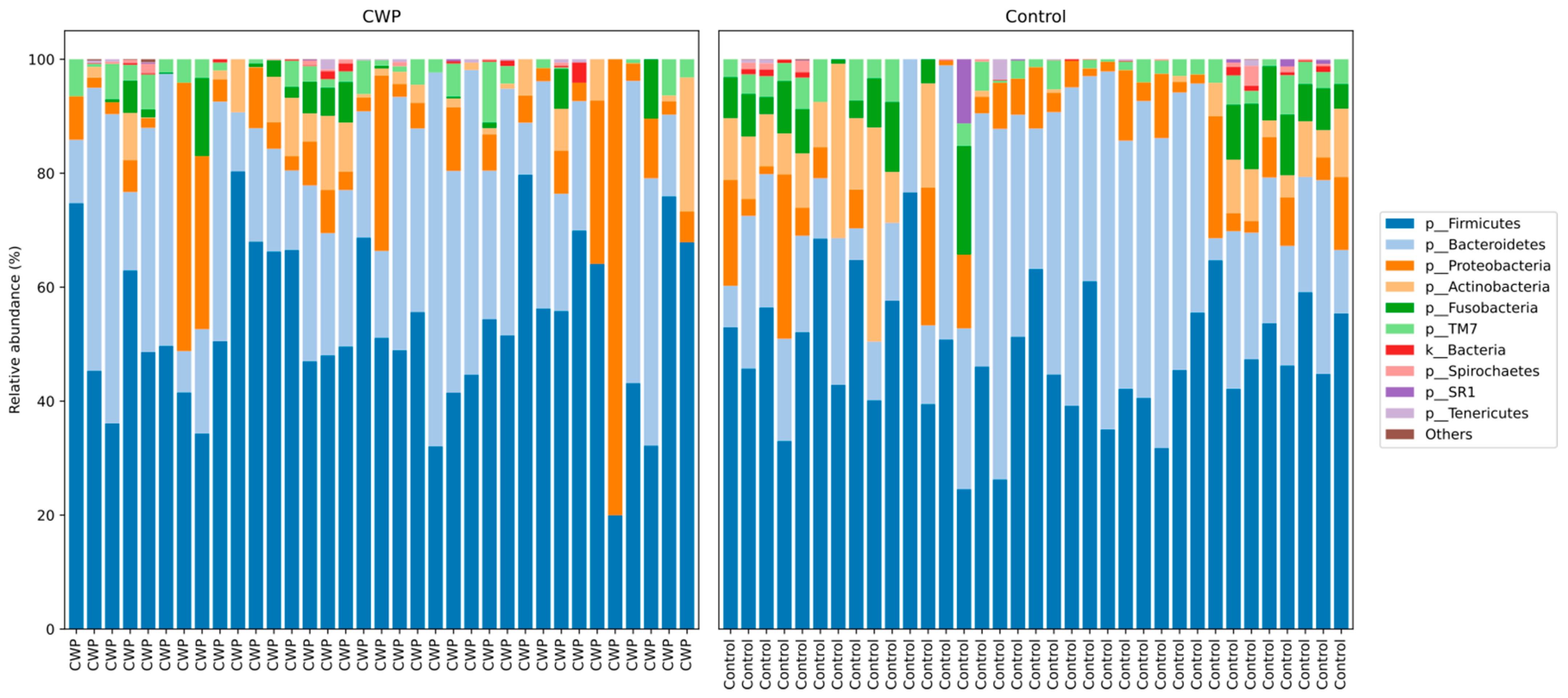
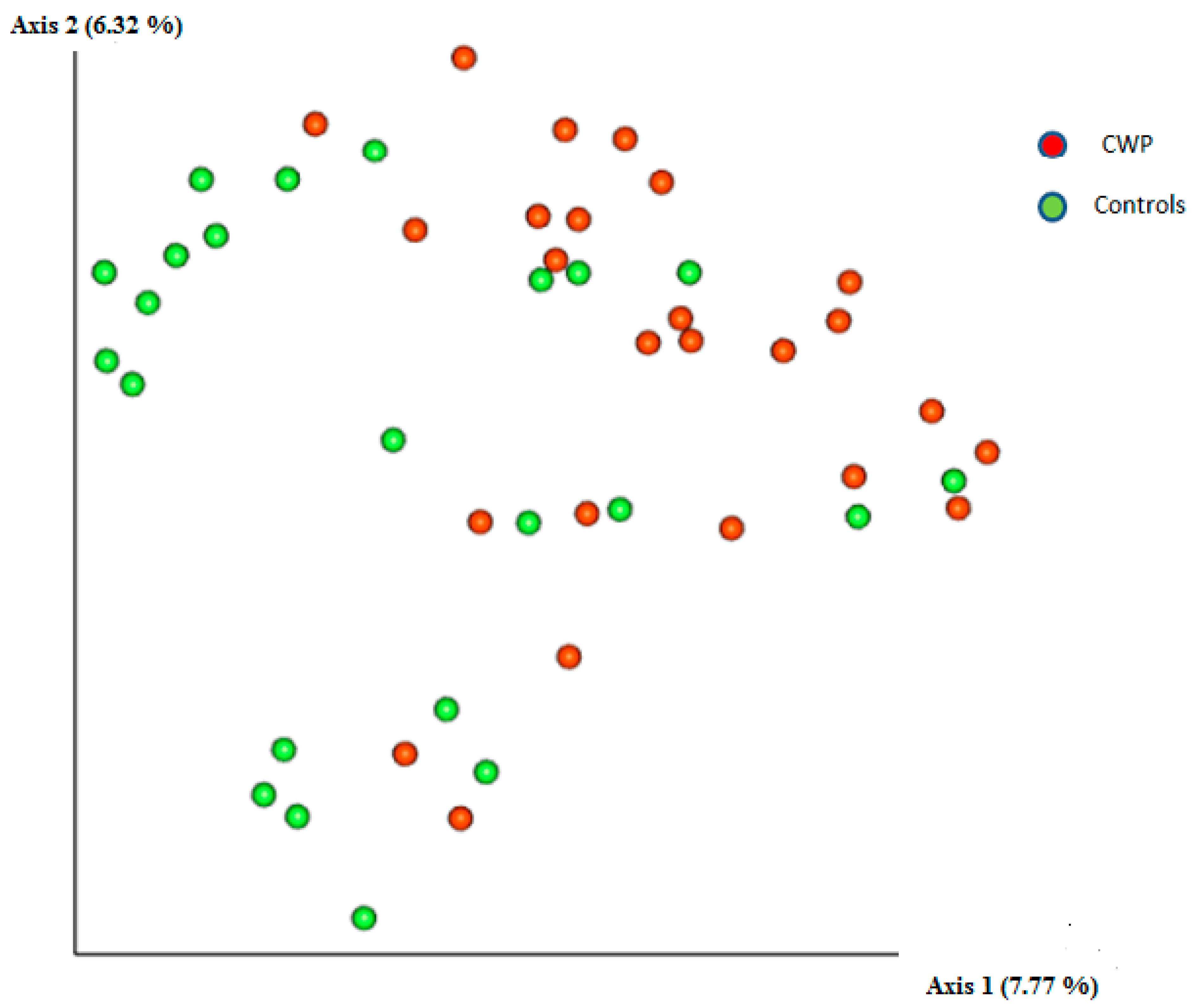
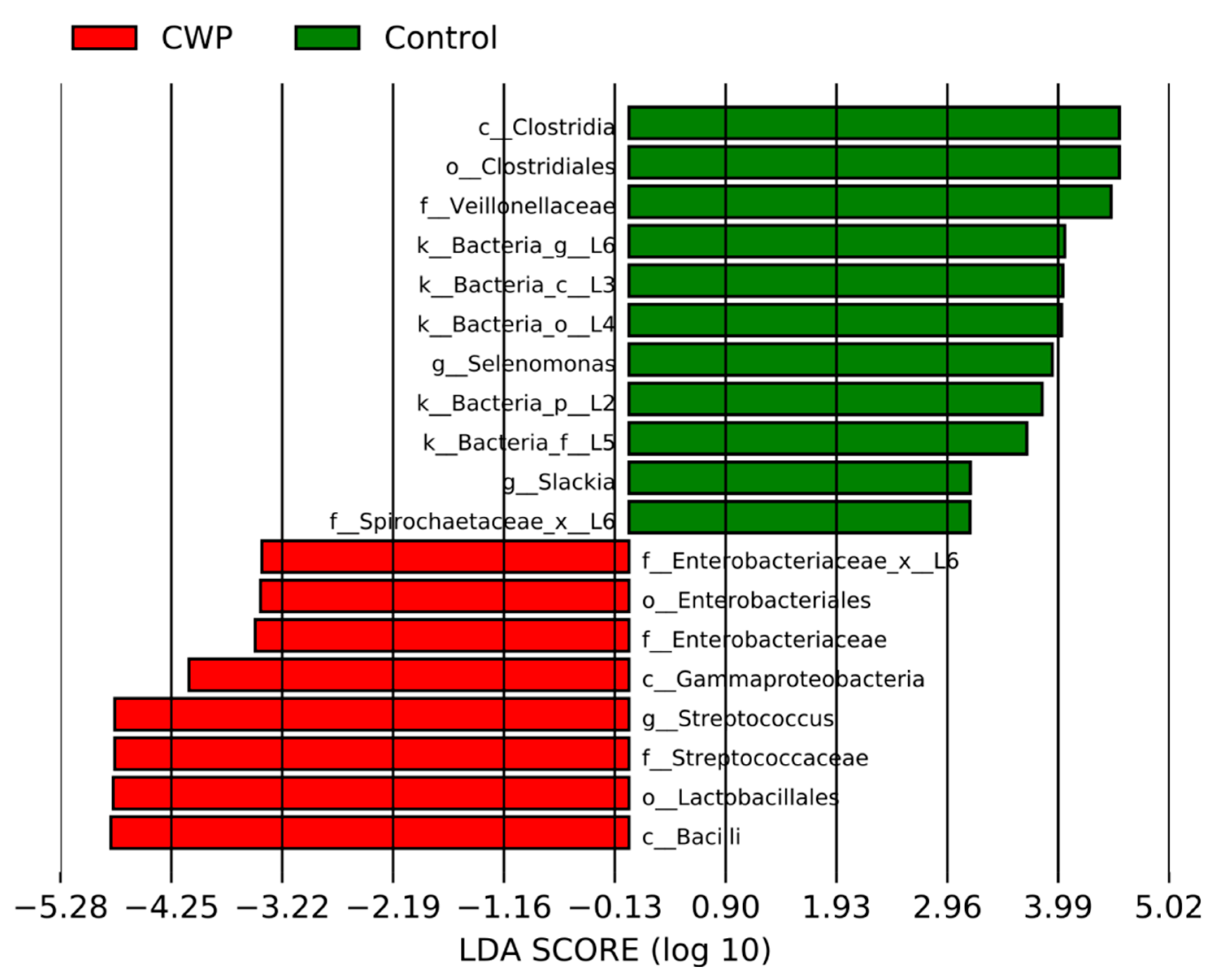
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, S.; Chen, H.; Harvey, M.A.; Stemn, E.; Cliff, D. Focusing on Coal Workers’ Lung Diseases: A Comparative Analysis of China, Australia, and the United States. Int. J. Environ. Res. Public Health 2018, 15, 2565. [Google Scholar] [CrossRef]
- Blackley, D.J.; Halldin, C.N.; Laney, A.S. Continued Increase in Prevalence of Coal Workers’ Pneumoconiosis in the United States, 1970–2017. Am. J. Public Health 2018, 108, 1220–1222. [Google Scholar] [CrossRef]
- Leonard, R.; Zulfikar, R.; Stansbury, R. Coal mining and lung disease in the 21st century. Curr. Opin. Pulm. Med. 2020, 26, 135–141. [Google Scholar] [CrossRef]
- Beer, C.; Kolstad, H.A.; Søndergaard, K.; Bendstrup, E.; Heederik, D.; Olsen, K.E.; Omland, Ø.; Petsonk, E.; Sigsgaard, T.; Sherson, D.L.; et al. A systematic review of occupational exposure to coal dust and the risk of interstitial lung diseases. Eur. Clin. Respir. J. 2017, 4, 1264711. [Google Scholar] [CrossRef] [PubMed]
- Tomaskova, H.; Jirak, Z.; Splichalova, A.; Urban, P. Cancer incidence in Czech black coal miners in association with coalworkers’ pneumoconiosis. Int. J. Occup. Med. Environ. Health 2012, 25, 137–1344. [Google Scholar] [CrossRef] [PubMed]
- Volobaev, V.P.; Sinitsky, M.Y.; Larionov, A.V.; Druzhinin, V.G.; Gafarov, N.I.; Minina, V.I.; Kulemin, J.E. Modifying influence of occupational inflammatory diseases on the level of chromosome aberrations in coal miners. Mutagenesis 2016, 31, 225–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Druzhinin, V.G.; Apalko, S.V.; Baranova, E.D.; Volobaev, V.P.; Drobchik, T.Y.; Larionov, A.V.; Hill, E.G.; Chasovskikh, E.V. Micronuclei in blood lumphocytes of existing and former coal miners: Evaluation of the effect of anthracosilicosis. Ecol. Genet. 2019, 17, 55–62. (In Russian) [Google Scholar] [CrossRef]
- Zhang, N.; Liu, K.; Wang, K.; Zhou, C.; Wang, H.; Che, S.; Liu, Z.; Yang, H. Dust induces lung fibrosis through dysregulated DNA methylation. Environ. Toxicol. 2019, 34, 728–741. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef]
- Caverly, L.J.; Huang, Y.J.; Sze, M.A. Past, Present, and Future Research on the Lung Microbiome in Inflammatory Airway Disease. Chest 2019, 156, 376–382. [Google Scholar] [CrossRef]
- Qi, Y.J.; Sun, X.J.; Wang, Z.; Bin, Y.F.; Li, Y.H.; Zhong, X.N.; Bai, J.; Deng, J.M.; He, Z.Y. Richness of sputum microbiome in acute exacerbations of eosinophilic chronic obstructive pulmonary disease. Chin. Med. J. 2020, 133, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.J.; Huang, Y.J. The microbiome in asthma: Role in pathogenesis, phenotype, and response to treatment. Ann. Allergy Asthma Immunol. 2019, 122, 270–275. [Google Scholar] [CrossRef]
- Wootton, D.G.; Cox, M.J.; Gloor, G.B.; Litt, D.; Hoschler, K.; German, E.; Court, J.; Eneje, O.; Keogan, L.; Macfarlane, L.; et al. A Haemophilus sp. dominates the microbiota of sputum from UK adults with non-severe community acquired pneumonia and chronic lung disease. Sci. Rep. 2019, 9, 2388. [Google Scholar] [CrossRef] [PubMed]
- Acosta, N.; Heirali, A.; Somayaji, R.; Surette, M.G.; Workentine, M.L.; Sibley, C.D.; Rabin, H.R.; Parkins, M.D. Sputum microbiota is predictive of long-term clinical outcomes in young adults with cystic fibrosis. Thorax 2018, 73, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Maddi, A.; Sabharwal, A.; Violante, T.; Manuballa, S.; Genco, R.; Patnaik, S.; Yendamuri, S. The microbiome and lung cancer. J. Thorac. Dis. 2019, 11, 280–291. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Molyneaux, P.L. The respiratory microbiome in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2017, 5, 250. [Google Scholar] [CrossRef]
- Jacob, N.; Jacobs, J.P.; Kumagai, K.; Ha, C.W.Y.; Kanazawa, Y.; Lagishetty, V.; Altmayer, K.; Hamill, A.M.; Von Arx, A.; Sartor, R.B.; et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 2018, 11, 1466–1476. [Google Scholar] [CrossRef]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef]
- Moghadamrad, S.; Hassan, M.; McCoy, K.D.; Kirundi, J.; Kellmann, P.; De Gottardi, A. Attenuated fibrosis in specific pathogen-free microbiota in experimental cholestasis- and toxin-induced liver injury. FASEB J. 2019, 33, 12464–12476. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 27, F1000 Faculty Rev-921. [Google Scholar] [CrossRef]
- Tagliari, E.; Campos, L.F.; Campos, A.C.; Costa-Casagrande, T.A.; Noronha, L. Effect of probiotic oral administration on skin wound healing in raTS. Arq. Bras. Cir. Dig. 2019, 32, e1457. [Google Scholar] [CrossRef] [PubMed]
- Buchta Rosean, C.; Bostic, R.R.; Ferey, J.C.M.; Feng, T.Y.; Azar, F.N.; Tung, K.S.; Dozmorov, M.G.; Smirnova, E.; Bos, P.D.; Rutkowski, M.R. Preexisting Commensal Dysbiosis Is a Host-Intrinsic Regulator of Tissue Inflammation and Tumor Cell Dissemination in Hormone Receptor-Positive Breast Cancer. Cancer Res. 2019, 79, 3662–3675. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ding, C.; Dai, X.; Lv, T.; Xie, T.; Zhang, T.; Gao, W.; Gong, J.; Zhu, W.; Li, N.; et al. Soluble Dietary Fiber Ameliorates Radiation-Induced Intestinal Epithelial-to-Mesenchymal Transition and Fibrosis. JPEN J. Parenter. Enteral. Nutr. 2017, 41, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef]
- Cameron, S.J.S.; Lewis, K.E.; Huws, S.A.; Hegarty, M.J.; Lewis, P.D.; Pachebat, J.A.; Mur, L.A.J. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS ONE 2017, 12, e0177062. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, L.; Sun, G.; Li, Y.; Huang, R. Alterations in the gut microbiota of patients with silica-induced pulmonary fibrosis. J. Occup. Med. Toxicol. 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.D. Molecular epidemiology of Streptococcus agalactiae (group B Streptococcus). Front. Biosci. 2003, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, L.L.; Chang, B.J.; Payne, M.S. Perinatal Streptococcus agalactiae Epidemiology and Surveillance Targets. Clin. Microbiol. Rev. 2018, 31, e00049-18. [Google Scholar] [CrossRef]
- Pimentel, B.A.; Martins, C.A.; Mendonça, J.C.; Miranda, P.S.; Sanches, G.F.; Mattos-Guaraldi, A.L.; Nagao, P.E. Streptococcus agalactiae infection in cancer patients: A five-year study. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Phares, C.R.; Lynfield, R.; Farley, M.M.; Mohle-Boetani, J.; Harrison, L.H.; Petit, S.; Craig, A.S.; Schaffner, W.; Zansky, S.M.; Gershman, K.; et al. Active Bacterial Core surveillance/Emerging Infections Program Network. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA 2008, 299, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Farley, M.M.; Harvey, R.C.; Stull, T.; Smith, J.D.; Schuchat, A.; Wenger, J.D.; Stephens, D.S. A population-based assessment of invasive disease due to group B Streptococcus in nonpregnant adults. N. Engl. J. Med. 1993, 328, 1807–1811. [Google Scholar] [CrossRef]
- Sharma, P.; Lata, H.; Arya, D.K.; Kashyap, A.K.; Kumar, H.; Dua, M.; Ali, A.; Johri, A.K. Role of pilus proteins in adherence and invasion of Streptococcus agalactiae to the lung and cervical epithelial cells. J. Biol. Chem. 2013, 288, 4023–4034. [Google Scholar] [CrossRef]
- da Costa, A.F.E.; Moraes, J.A.; de Oliveira, J.S.S.; Dos Santos, M.H.B.; da Silva Santos, G.; Barja-Fidalgo, C.; Mattos-Guaraldi, A.L.; Nagao, P.E. Reactive oxygen species involved in apoptosis induction of human respiratory epithelial (A549) cells by Streptococcus agalactiae. Microbiology 2016, 162, 94–99. [Google Scholar] [CrossRef]
- Oliveira, J.S.S.; Santos, G.D.S.; Moraes, J.A.; Saliba, A.M.; Barja-Fidalgo, T.C.; Mattos-Guaraldi, A.L.; Nagao, P.E. Reactive oxygen species generation mediated by NADPH oxidase and PI3K/Akt pathways contribute to invasion of Streptococcus agalactiae in human endothelial cells. Mem. Inst. Oswaldo Cruz. 2018, 113, e140421. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. COMET Investigators. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef]
- Skolnik, K.; Nguyen, A.; Thornton, C.S.; Waddell, B.; Williamson, T.; Rabin, H.R.; Parkins, M.D. Group B streptococcus (GBS) is an important pathogen in human disease- but what about in cystic fibrosis? BMC Infect. Dis. 2017, 17, 660. [Google Scholar] [CrossRef]
- Pragman, A.A.; Kim, H.B.; Reilly, C.S.; Wendt, C.; Isaacson, R.E. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS ONE 2012, 7, e47305. [Google Scholar] [CrossRef]
- Einarsson, G.G.; Comer, D.M.; McIlreavey, L.; Parkhill, J.; Ennis, M.; Tunney, M.M.; Elborn, J.S. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax 2016, 71, 795–803. [Google Scholar] [CrossRef]
- Cabrera-Rubio, R.; Garcia-Núñez, M.; Setó, L.; Antó, J.M.; Moya, A.; Monsó, E.; Mira, A. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J. Clin. Microbiol. 2012, 50, 3562–3568. [Google Scholar] [CrossRef]
- Haldar, K.; George, L.; Wang, Z.; Mistry, V.; Ramsheh, M.Y.; Free, R.C.; John, C.; Reeve, N.F.; Miller, B.E.; Tal-Singer, R.; et al. The sputum microbiome is distinct between COPD and health, independent of smoking history. Respir. Res. 2020, 21, 183. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Wang, F.; Yang, Y.; Wang, X.; Chen, B.; Stampfli, M.R.; Zhou, H.; Shu, W.; Brightling, C.E.; et al. A Refined View of Airway Microbiome in Chronic Obstructive Pulmonary Disease at Species and Strain-Levels. Front. Microbiol. 2020, 30, 1758. [Google Scholar] [CrossRef]
- Druzhinin, V.G.; Matskova, L.V.; Demenkov, P.S.; Baranova, E.D.; Volobaev, V.P.; Minina, V.I.; Larionov, A.V.; Titov, V.A.; Fucic, A. Genetic damage in lymphocytes of lung cancer patients is correlated to the composition of the respiratory tract microbiome. Mutagenesis 2021, 36, 143–153. [Google Scholar] [CrossRef]
- Liu, H.X.; Tao, L.L.; Zhang, J.; Zhu, Y.G.; Zheng, Y.; Liu, D.; Zhou, M.; Ke, H.; Shi, M.M.; Qu, J.M. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int. J. Cancer. 2018, 142, 769–778. [Google Scholar] [CrossRef]
- Hosgood, H.D., 3rd; Sapkota, A.R.; Rothman, N.; Rohan, T.; Hu, W.; Xu, J.; Vermeulen, R.; He, X.; White, J.R.; Wu, G.; et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environ. Mol. Mutagen. 2014, 55, 643–651. [Google Scholar] [CrossRef]
- Ran, Z.; Liu, J.; Wang, F.; Xin, C.; Shen, X.; Zeng, S.; Song, Z.; Xiong, B. Analysis of Pulmonary Microbial Diversity in Patients with Advanced Lung Cancer Based on High-throughput Sequencing Technology. Chin. J. Lung Cancer 2020, 23, 1031–1038. (In Chinese) [Google Scholar] [CrossRef]
- Hasegawa, A.; Sato, T.; Hoshikawa, Y.; Ishida, N.; Tanda, N.; Kawamura, Y.; Kondo, T.; Takahashi, N. Detection and identification of oral anaerobes in intraoperative bronchial fluids of patients with pulmonary carcinoma. Microbiol. Immunol. 2014, 58, 375–381. [Google Scholar] [CrossRef]
- Liu, Y.; O’Brien, J.L.; Ajami, N.J.; Scheurer, M.E.; Amirian, E.S.; Armstrong, G.; Tsavachidis, S.; Thrift, A.P.; Jiao, L.; Wong, M.C.; et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am. J. Cancer Res. 2018, 8, 1775–1787. [Google Scholar]
- Zhang, W.; Luo, J.; Dong, X.; Zhao, S.; Hao, Y.; Peng, C.; Shi, H.; Zhou, Y.; Shan, L.; Sun, Q.; et al. Salivary Microbial Dysbiosis is Associated with Systemic Inflammatory Markers and Predicted Oral Metabolites in Non-Small Cell Lung Cancer Patients. J. Cancer 2019, 10, 1651–1662. [Google Scholar] [CrossRef]
- Tsay, J.J.; Wu, B.G.; Badri, M.H.; Clemente, J.C.; Shen, N.; Meyn, P.; Li, Y.; Yie, T.A.; Lhakhang, T.; Olsen, E.; et al. Airway Microbiota Is Associated with Upregulation of the PI3K Pathway in Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 1188–1198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variables | Coal Worker’s Pneumoconiosis, n = 35 | Healthy Kemerovo Residents (Control), n = 35 |
|---|---|---|
| Age (years) (mean ± SD) | 58.5 ± 8.3 | 55.7 ± 11.7 |
| Stage of CWP (%): | - | |
| I | 88.6 | |
| II | 11.4 | |
| Respiratory symptoms (%): | ||
| Cough | 22.9 | 8.6 |
| Dyspnea | 5.7 | 5.7 |
| Chronic diseases (%): | ||
| Heart and vessels | 34.3 | 34.3 |
| Bronchitis | 14.3 | 5.7 |
| Stomach | 14.3 | 11.4 |
| Diabetes | 5.7 | 5.7 |
| Obesity | 5.7 | 2.9 |
| Living environment (%): | ||
| City | 94.3 | 91.4 |
| Village | 5.7 | 8.6 |
| Coal dust occupational exposure (%): | ||
| Current miners | 23.0 | |
| Former miners | 77.0 | - |
| Smoking status (%): | ||
| Non-smokers | 97.1 | 94.3 |
| Smokers | 2.9 | 5.7 |
| Alcohol status (%): | ||
| Non-drinker | 14.3 | 20.0 |
| Rare drinker | 77.1 | 48.5 |
| Medium drinker | 8.6 | 31.5 |
| Diet (%): | ||
| Vegetarian | 0 | 0 |
| Non-vegetarian | 100 | 100 |
| Phyla | Controls, Mean ± SD | CWP, Mean ± SD | p Value |
|---|---|---|---|
| Firmicutes | 47.46 ± 11.25 | 50.41 ± 14.1 | 0.39 |
| Bacteroidetes | 22.93 ± 10.88 | 19.83 ± 11.51 | 0.29 |
| Actinobacteria | 10.1 ± 7.91 | 8.99 ± 5.76 | 0.85 |
| Proteobacteria | 8.24 ± 6.54 | 11.45 ± 16.56 | 0.9 |
| Fusobacteria | 6.98 ± 4.45 | 4.56 ± 3.96 | 0.02 |
| TM7 | 1.8 ± 1.68 | 1.53 ± 1.84 | 0.44 |
| Spirochaetes | 0.53 ± 0.96 | 0.6 ± 0.99 | 0.53 |
| Tenericutes | 0.18 ± 0.74 | 0.23 ± 0.41 | 0.1 |
| Genus | Controls | CWP | p Value | p Value (FDR) |
|---|---|---|---|---|
| Mean ± SD | Mean ± SD | |||
| Streptococcus | 16.85 ± 11.35 | 25.12 ± 11.37 | 0.0003 * | 0.0023 |
| Prevotella (f. Prevotellfceae) | 15.36 ± 7.83 | 13.24 ± 8.06 | >0.05 | 0.0114 |
| Veillonella | 15.4 ± 11.82 | 12.03 ± 10.14 | >0.05 | 0.0136 |
| Anaerosinus | 15.36 ± 11.86 | 11.56 ± 10.18 | >0.05 | 0.0159 |
| Selenomonas | 4.6 ± 4.39 | 2.07 ± 2.1 | 0.02 | 0.0045 |
| Porphyromonas | 3.28 ± 3.5 | 3.26 ± 5.84 | >0.05 | 0.0182 |
| Actinomyces | 5.95 ± 6.75 | 4.24 ± 3.23 | >0.05 | 0.0205 |
| Megasphaera | 2.46 ± 1.43 | 1.45 ± 1.6 | >0.05 | 0.0227 |
| Alloprevotella | 2.2 ± 2.29 | 2.78 ± 2.92 | >0.05 | 0.0250 |
| Streptobacillus | 2.98 ± 2.99 | 2.3 ± 2.88 | >0.05 | 0.0273 |
| Leptotrichia | 3.04 ± 3.01 | 2.34 ± 2.74 | >0.05 | 0.0295 |
| Granulicatella | 1.01 ± 1.39 | 1.71 ± 1.68 | 0.03 | 0.0068 |
| Gemella | 2.03 ± 1.93 | 3.12 ± 2.19 | >0.05 | 0.0318 |
| Rothia | 2.27 ± 3.02 | 2.65 ± 3.05 | >0.05 | 0.0341 |
| Bacillus | 1.67 ± 1.7 | 2.67 ± 2.19 | 0.04 | 0.0091 |
| Atopobium | 1.41 ± 1.91 | 1.43 ± 1.39 | >0.05 | 0.0364 |
| Pasteurellaceae | 2.34 ± 1.21 | 3.27 ± 2.35 | >0.05 | 0.0386 |
| Fusobacterium | 1.91 ± 1.82 | 1.65 ± 1.42 | >0.05 | 0.0409 |
| Macellibacteroides | 2.01 ± 2.58 | 1.37 ± 2.58 | >0.05 | 0.0432 |
| Neisseria | 3.83 ± 4.91 | 4.72 ± 12.27 | >0.05 | 0.0454 |
| Bacteroides | 1.15 ± 1.6 | 0.82 ± 1.78 | >0.05 | 0.0477 |
| Prevotella (f. Paraprevotellacacea) | 1.17 ± 1.69 | 0.99 ± 1.41 | >0.05 | 0.0500 |
| Species | Controls | CWP | p Value | p Value (FDR) |
|---|---|---|---|---|
| Mean ± SD | Mean ± SD | |||
| Streptococcus agalactiae | 16.93 ± 10.92 | 25.32 ± 11.55 | 0.0002 * | 0.0016 |
| Anaerosinus glycerini | 14.27 ± 12.03 | 11.5 ± 10.22 | >0.05 | 0.0048 |
| Selenomonas bovis | 4.28 ± 4.28 | 1.98 ± 1.99 | 0.03 | 0.0032 |
| Megasphaera micronuciformis | 2.35 ± 2.87 | 1.66 ± 1.71 | >0.05 | 0.0065 |
| Prevotella histicola | 1.44 ± 1.92 | 2.64 ± 3.33 | >0.05 | 0.0081 |
| Actinomyces hyovaginalis | 4.51 ± 5.78 | 2.19 ± 1.67 | >0.05 | 0.0097 |
| Granulicatella balaenopterae | 1.63 ± 1.54 | 1.69 ± 1.66 | >0.05 | 0.0113 |
| Atopobium rimae | 1.41 ± 1.91 | 1.44 ± 1.38 | >0.05 | 0.0129 |
| Prevotella pallens | 1.54 ± 1.96 | 0.92 ± 1.74 | >0.05 | 0.0145 |
| Rothia terrae | 1.84 ± 2.56 | 2.44 ± 2.97 | >0.05 | 0.0161 |
| Macellibacteroides fermentans | 1.93 ± 2.43 | 1.38 ± 2.59 | >0.05 | 0.0177 |
| Bacteroides nordii | 1.25 ± 1.58 | 0.75 ± 1.56 | >0.05 | 0.0194 |
| Prevotella tannarae | 0.52 ± 1.08 | 0.7 ± 1.27 | >0.05 | 0.0210 |
| Lachnoanaerobaculum orale | 0.56 ± 0.74 | 0.55 ± 0.63 | >0.05 | 0.0226 |
| Prevotella intermedia | 0.44 ± 1.01 | 0.48 ± 0.7 | >0.05 | 0.0242 |
| Prevotella nigrescens | 0.37 ± 0.61 | 0.22 ± 0.39 | >0.05 | 0.0258 |
| Prevotella nanceiensis | 0.61 ± 1.09 | 0.43 ± 0.58 | >0.05 | 0.0274 |
| Bulleidia moorei | 0.23 ± 0.3 | 0.29 ± 0.43 | >0.05 | 0.0290 |
| Clostridium bolteae | 0.22 ± 0.5 | 0.32 ± 0.56 | >0.05 | 0.0306 |
| Porphyromonas endodontalis | 0.67 ± 1.42 | 1.49 ± 5.07 | >0.05 | 0.0323 |
| Vestibaculum illigatum | 0.5 ± 1.27 | 0.31 ± 0.65 | >0.05 | 0.0339 |
| Clostridium acidurici | 0.27 ± 0.8 | 0.26 ± 0.73 | >0.05 | 0.0355 |
| Mycoplasma zalophi | 0.3 ± 0.87 | 0.29 ± 0.43 | >0.05 | 0.0371 |
| Moryella indoligenes | 0.27 ± 0.64 | 0.2 ± 0.53 | >0.05 | 0.0387 |
| Peptostreptococcus anaerobius | 0.2 ± 0.54 | 0.23 ± 0.49 | >0.05 | 0.0403 |
| Treponema amulovorum | 0.11 ± 0.24 | 0.07 ± 0.22 | >0.05 | 0.0419 |
| Oribacterium sinus | 0.39 ± 0.61 | 0.13 ± 0.27 | >0.05 | 0.0435 |
| Leptotrichia trevisanii | 0.1 ± 0.25 | 0.14 ± 0.31 | > 0.05 | 0.0452 |
| Lactobacillus hamsteri | 0.09 ± 0.43 | 0.11 ± 0.62 | >0.05 | 0.0468 |
| Bergeyella zoohelcum | 0.32 ± 0.78 | 0.13 ± 0.2 | >0.05 | 0.0484 |
| Campylobacter rectus | 0.08 ± 0.2 | 0.11 ± 0.24 | >0.05 | 0.0500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Druzhinin, V.G.; Baranova, E.D.; Matskova, L.V.; Demenkov, P.S.; Volobaev, V.P.; Minina, V.I.; Larionov, A.V.; Paradnikova, S.A. Sputum Microbiota in Coal Workers Diagnosed with Pneumoconiosis as Revealed by 16S rRNA Gene Sequencing. Life 2022, 12, 830. https://doi.org/10.3390/life12060830
Druzhinin VG, Baranova ED, Matskova LV, Demenkov PS, Volobaev VP, Minina VI, Larionov AV, Paradnikova SA. Sputum Microbiota in Coal Workers Diagnosed with Pneumoconiosis as Revealed by 16S rRNA Gene Sequencing. Life. 2022; 12(6):830. https://doi.org/10.3390/life12060830
Chicago/Turabian StyleDruzhinin, Vladimir G., Elizaveta D. Baranova, Ludmila V. Matskova, Pavel S. Demenkov, Valentin P. Volobaev, Varvara I. Minina, Alexey V. Larionov, and Snezana A. Paradnikova. 2022. "Sputum Microbiota in Coal Workers Diagnosed with Pneumoconiosis as Revealed by 16S rRNA Gene Sequencing" Life 12, no. 6: 830. https://doi.org/10.3390/life12060830
APA StyleDruzhinin, V. G., Baranova, E. D., Matskova, L. V., Demenkov, P. S., Volobaev, V. P., Minina, V. I., Larionov, A. V., & Paradnikova, S. A. (2022). Sputum Microbiota in Coal Workers Diagnosed with Pneumoconiosis as Revealed by 16S rRNA Gene Sequencing. Life, 12(6), 830. https://doi.org/10.3390/life12060830