
Review of Multiple Myeloma Genetics including Effects on Prognosis, Response to Treatment, and Diagnostic Workup


Abstract
:1. Introduction
2. Genetic Aberrations in Multiple Myeloma
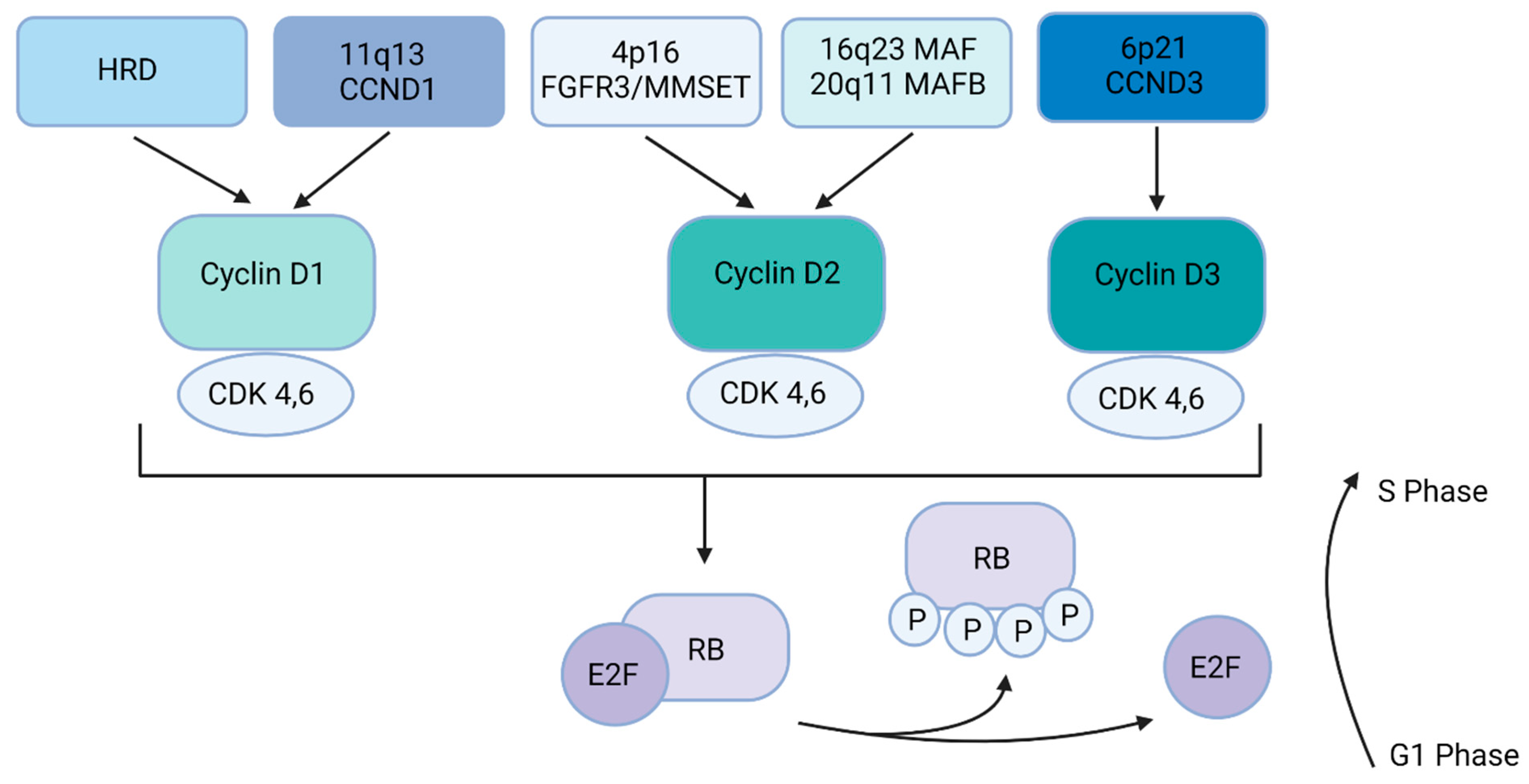
2.1. Primary or Clonal Genetic Events in Multiple Myeloma
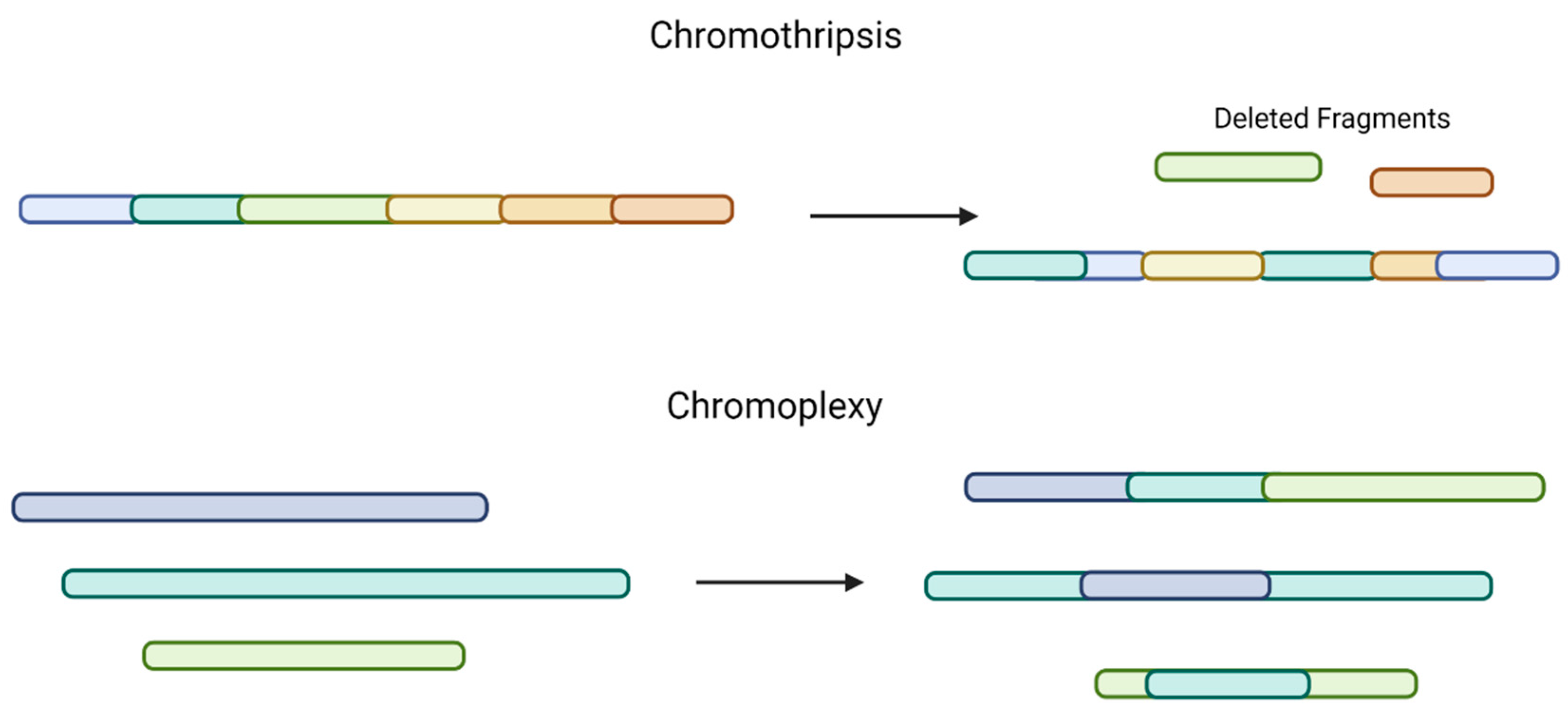
2.2. Secondary or Sub-Clonal Genetic Events in Multiple Myeloma
2.3. Evolution and Spatial Heterogeneity of MM, Based on Genetics
3. Prognosis in MM and Precursor Conditions, Based on Genetic Aberrations
4. Current Molecular Diagnostic Workup for MM, including FISH, NGS, and Mass Spectrometry
5. The Future of Genomics for MM: Workup and Treatment
6. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J. Clin. Oncol. 2020, 38, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef]
- Fonseca, R.; Barlogie, B.; Bataille, R.; Bastard, C.; Bergsagel, P.L.; Chesi, M.; Davies, F.E.; Drach, J.; Greipp, P.R.; Kirsch, I.R.; et al. Genetics and cytogenetics of multiple myeloma: A workshop report. Cancer Res. 2004, 64, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Corre, J.; Avet-Loiseau, H. Risk Stratification and Targets in Multiple Myeloma: From Genomics to the Bedside. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 675–680. [Google Scholar] [CrossRef]
- Shaughnessy, J., Jr.; Gabrea, A.; Qi, Y.; Brents, L.; Zhan, F.; Tian, E.; Sawyer, J.; Barlogie, B.; Bergsagel, P.L.; Kuehl, M. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood 2001, 98, 217–223. [Google Scholar] [CrossRef]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schröck, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef]
- Chesi, M.; Bergsagel, P.L.; Shonukan, O.O.; Martelli, M.L.; Brents, L.A.; Chen, T.; Schröck, E.; Ried, T.; Kuehl, W.M. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 1998, 91, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Nardini, E.; Lim, R.S.; Smith, K.D.; Kuehl, W.M.; Bergsagel, P.L. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998, 92, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Hanamura, I.; Iida, S.; Akano, Y.; Hayami, Y.; Kato, M.; Miura, K.; Harada, S.; Banno, S.; Wakita, A.; Kiyoi, H.; et al. Ectopic expression of MAFB gene in human myeloma cells carrying (14;20)(q32;q11) chromosomal translocations. Jpn. J. Cancer Res. 2001, 92, 638–644. [Google Scholar] [CrossRef]
- Morgan, G.; Walker, B.; Davies, F. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Barilà, G.; Bonaldi, L.; Grassi, A.; Martines, A.; Liço, A.; Macrì, N.; Nalio, S.; Pavan, L.; Berno, T.; Branca, A.; et al. Identification of the true hyperdiploid multiple myeloma subset by combining conventional karyotyping and FISH analysis. Blood Cancer J. 2020, 10, 18. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Li, C.; Magrangeas, F.; Gouraud, W.; Charbonnel, C.; Harousseau, J.L.; Attal, M.; Marit, G.; Mathiot, C.; Facon, T.; et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 2009, 27, 4585–4590. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.M.; Fonseca, R.; Usmani, S.Z. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021, 11, 83. [Google Scholar] [CrossRef]
- Raab, M.S.; Giesen, N.; Scheid, C.; Besemer, B.; Miah, K.; Benner, A.; Metzler, I.; Khandanpour, C.; Seidel-Glaetzer, A.; Trautmann-Grill, K.; et al. Safety and Preliminary Efficacy Results from a Phase II Study Evaluating Combined BRAF and MEK Inhibition in Relapsed/Refractory Multiple Myeloma (rrMM) Patients with Activating BRAF V600E Mutations: The GMMG-Birma Trial. Blood 2020, 136, 44–45. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [Green Version]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.G.; Neri, P.; Bahlis, N.J.; Nooka, A.K.; Dhodapkar, M.V.; Jaye, D.L.; Hofmeister, C.C.; Kaufman, J.L.; Gupta, V.A.; Auclair, D.; et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat. Commun. 2019, 10, 1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Promiscuous Structural Variants Drive Myeloma Initiation and Progression. Blood Cancer Discov. 2020, 1, 221–223. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Facon, T.; Grosbois, B.; Magrangeas, F.; Rapp, M.-J.; Harousseau, J.-L.; Minvielle, S.; Bataille, R. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood 2002, 99, 2185–2191. [Google Scholar] [CrossRef]
- Fonseca, R.; Oken, M.M.; Greipp, P.R. Eastern Cooperative Oncology Group Myeloma Group. The t(4;14)(p16.3;q32) is strongly associated with chromosome 13 abnormalities in both multiple myeloma and monoclonal gammopathy of undetermined significance. Blood 2001, 98, 1271–1272. [Google Scholar] [CrossRef]
- Diamond, B.; Yellapantula, V.; Rustad, E.H.; Maclachlan, K.H.; Mayerhoefer, M.; Kaiser, M.; Morgan, G.; Landgren, O.; Maura, F. Positive selection as the unifying force for clonal evolution in multiple myeloma. Leukemia 2021, 35, 1511–1515. [Google Scholar] [CrossRef]
- Paiva, B.; Paino, T.; Sayagues, J.-M.; Garayoa, M.; San-Segundo, L.; Martín, M.; Mota, I.; Sanchez, M.-L.; Bárcena, P.; Aires-Mejia, I.; et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood 2013, 122, 3591–3598. [Google Scholar] [CrossRef] [Green Version]
- Foulk, B.; Schaffer, M.; Gross, S.; Rao, C.; Smirnov, D.; Connelly, M.C.; Chaturvedi, S.; Reddy, M.; Brittingham, G.; Mata, M.; et al. Enumeration and characterization of circulating multiple myeloma cells in patients with plasma cell disorders. Br. J. Haematol. 2018, 180, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcés, J.-J.; Simicek, M.; Vicari, M.; Brozova, L.; Burgos, L.; Bezdekova, R.; Alignani, D.; Calasanz, M.-J.; Growkova, K.; Goicoechea, I.; et al. Transcriptional profiling of circulating tumor cells in multiple myeloma: A new model to understand disease dissemination. Leukemia 2020, 34, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; van Ness, B.; van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas—Current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef]
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.; van Wier, S.A.; Chng, W.J.; Ketterling, R.; Lacy, M.Q.; Dispenzieri, A.; Bergsagel, P.L.; Rajkumar, S.V.; Greipp, P.R.; Litzow, M.R.; et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006, 20, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. VISTA Trial Investigators. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906. [Google Scholar] [CrossRef] [Green Version]
- Giri, S.; Huntington, S.F.; Wang, R.; Zeidan, A.M.; Podoltsev, N.; Gore, S.D.; Ma, X.; Gross, C.P.; Davidoff, A.J.; Neparidze, N. Chromosome 1 abnormalities and survival of patients with multiple myeloma in the era of novel agents. Blood Adv. 2020, 4, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Hebraud, B.; Leleu, X.; Lauwers-Cances, V.; Roussel, M.; Caillot, D.; Marit, G.; Karlin, L.; Hulin, C.; Gentil, C.; Guilhot, F.; et al. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: The IFM experience on 1195 patients. Leukemia 2014, 28, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesi, M.; Stein, C.K.; Garbitt, V.M.; Sharik, M.E.; Asmann, Y.W.; Bergsagel, M.; Riggs, D.L.; Welsh, S.J.; Meermeier, E.W.; Kumar, S.K.; et al. Monosomic loss of MIR15A/MIR16-1 is a driver of multiple myeloma proliferation and disease progression. Blood Cancer Discov. 2020, 1, 68–81. [Google Scholar] [CrossRef]
- Shou, Y.; Martelli, M.L.; Gabrea, A.; Qi, Y.; Brents, L.A.; Roschke, A.; Dewald, G.; Kirsch, I.R.; Bergsagel, P.L.; Kuehl, W.M. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc. Natl. Acad. Sci. USA 2000, 97, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Petljak, M.; Lionetti, M.; Cifola, I.; Liang, W.; Pinatel, E.M.; Alexandrov, L.B.; Fullam, A.; Martincorena, I.; Dawson, K.J.; et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2018, 32, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat. Commun. 2021, 12, 1861. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Murray, D.L.; Puig, N.; Kristinsson, S.; Usmani, S.Z.; Dispenzieri, A.; Bianchi, G.; Kumar, S.; Chng, W.J.; Hajek, R.; Paiva, B.; et al. Mass spectrometry for the evaluation of monoclonal proteins in multiple myeloma and related disorders: An International Myeloma Working Group Mass Spectrometry Committee Report. Blood Cancer J. 2021, 11, 24. [Google Scholar] [CrossRef]
- Paiva, B.; Vidriales, M.-B.; Cerveró, J.; Mateo, G.; Pérez, J.J.; Montalbán, M.A.; Sureda, A.; Montejano, L.; Gutiérrez, N.C.; de Coca, A.G.; et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood 2008, 112, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.D.L.; Vidriales, M.-B.; Pérez, J.J.; Mateo, G.; Montalbán, M.A.; Mateos, M.V.; Bladé, J.; Lahuerta, J.J.; Orfao, A.; Miguel, J.F.S. Multiparameter flow cytometry quantification of bone marrow plasma cells at diagnosis provides more prognostic information than morphological assessment in myeloma patients. Haematologica 2009, 94, 1599–1602. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; González, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.D.L.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A large meta-analysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta, J.-J.; Paiva, B.; Vidriales, M.-B.; Cordón, L.; Cedena, M.-T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.-L.; et al. Depth of response in multiple myeloma: A pooled analysis of three PETHEMA/GEM clinical trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: A meta-analysis. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Tschautscher, M.A.; Jevremovic, A.; Rajkumar, V.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Dingli, D.; Hwa, Y.L.; Fonder, A.L.; et al. Prognostic value of minimal residual disease and polyclonal plasma cells in myeloma patients achieving a complete response to therapy. Am. J. Hematol. 2019, 94, 751–756. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, E328–E346. [Google Scholar] [CrossRef]
- Maclachlan, K.H.; Came, N.; Diamond, B.; Roshal, M.; Ho, C.; Thoren, K.; Mayerhoefer, M.E.; Landgren, O.; Harrison, S. Minimal residual disease in multiple myeloma: Defining the role of next generation sequencing and flow cytometry in routine diagnostic use. Pathology 2021, 53, 385–399. [Google Scholar] [CrossRef]
- Medina, A.; Puig, N.; Flores-Montero, J.; Jimenez, C.; Sarasquete, M.-E.; Garcia-Alvarez, M.; Prieto-Conde, I.; Chillon, C.; Alcoceba, M.; Gutierrez, N.C.; et al. Comparison of next-generation sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma. Blood Cancer J. 2020, 10, 108. [Google Scholar] [CrossRef]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and prevalence of monoclonal gammopathy of undetermined significance: A study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellapantula, V.; Hultcrantz, M.; Rustad, E.H.; Wasserman, E.; Londono, D.; Cimera, R.; Ciardiello, A.; Landau, H.; Akhlaghi, T.; Mailankody, S.; et al. Comprehensive detection of recurring genomic abnormalities: A targeted sequencing approach for multiple myeloma. Blood Cancer J. 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Smadbeck, J.B.; Abdallah, N.; Zepeda-Mendoza, C.; Binder, M.; Pearce, K.E.; Asmann, Y.W.; Peterson, J.F.; Ketterling, R.P.; Greipp, P.T.; et al. The Prognostic Role of MYC Structural Variants Identified by NGS and FISH in Multiple Myeloma. Clin. Cancer Res. 2021, 27, 5430–5439. [Google Scholar] [CrossRef] [PubMed]
- Milanez-Almeida, P.; Martins, A.J.; Germain, R.N.; Tsang, J.S. Cancer prognosis with shallow tumor RNA sequencing. Nat. Med. 2020, 26, 188–192. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J.L.; Gasparetto, C.; Schjesvold, F.H.; Moreau, P.; Touzeau, C.; Facon, T.; Boise, L.H.; Jiang, Y.; Yang, X.; Dunbar, F.; et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am. J. Hematol. 2021, 96, 418–427. [Google Scholar] [CrossRef]
- Gupta, V.A.; Barwick, B.G.; Matulis, S.M.; Shirasaki, R.; Jaye, D.L.; Keats, J.J.; Oberlton, B.; Joseph, N.S.; Hofmeister, C.C.; Heffner, L.T.; et al. Venetoclax sensitivity in multiple myeloma is associated with B-cell gene expression. Blood 2021, 137, 3604–3615. [Google Scholar] [CrossRef]
- Slomp, A.; Moesbergen, L.M.; Gong, J.-N.; Cuenca, M.; von dem Borne, P.A.; Sonneveld, P.; Huang, D.C.S.; Minnema, M.C.; Peperzak, V. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019, 3, 4202–4214. [Google Scholar] [CrossRef]
- Vanderkerken, K.; de Veirman, K.; Maes, K.; Menu, E.; de Bruyne, E. MCL1 Inhibitors in Multiple Myeloma. Blood 2019, 134 (Suppl. 1), SCI-12. [Google Scholar] [CrossRef]
- Grand, E.K.; Chase, A.J.; Heath, C.; Rahemtulla, A.; Cross, N.C. Targeting FGFR3 in multiple myeloma: Inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia 2004, 18, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Chau, I.; Hyman, D.M.; Ribrag, V.; Blay, J.-Y.; Tabernero, J.; Elez, E.; Wolf, J.; Yee, A.J.; Kaiser, M.; et al. Vemurafenib in Patients with Relapsed Refractory Multiple Myeloma Harboring BRAFV600 Mutations: A Cohort of the Histology-Independent VE-BASKET Study. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rustad, E.; Dai, H.Y.; Hov, H.; Coward, E.; Beisvag, V.; Myklebost, O.; Hovig, E.; Nakken, S.; Vodák, D.; Meza-Zepeda, L.A.; et al. BRAF V600E mutation in early-stage multiple myeloma: Good response to broad acting drugs and no relation to prognosis. Blood Cancer J. 2015, 5, e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the initiation of multiple myeloma. Nat. Commun. 2020, 11, 1917. [Google Scholar] [CrossRef] [Green Version]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Kremer, R.; Cattoretti, G.; Bergsagel, P.L. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Primary Translocations | IgH Translocation Partner | Frequency |
|---|---|---|
| t(11;14) | Cyclin D1 (CCND1) | ~16% |
| t(4;14) | FGFR3/MMSET | ~15% |
| t(6;14) | Cyclin D3 (CCND3) | ~6% |
| t(4:16) | MAF | ~5% |
| t(14;20) | MAFB | ~2% |
| Copy Number Abnormality | Affected Genes | Frequency |
|---|---|---|
| Deletion 13q (del(13q)) | RB1, DIS3, MIR15A/MIR16 | ~45% |
| Gain 1q | MCL1 and CKS1B | ~40% |
| Deletion 14q (del(14q)) | TRAF3 | ~20% |
| Deletion 17 p (del(17p)) | TP53 | ~8% |
| Deletion del (1p) | CDKN2C | ~10% |
| Secondary Translocations | Translocation Partner | Frequency |
| Myc | Variable | ~25–50% |
| MAP3K14 | Variable | ~5% |
| Pathway | Genes |
|---|---|
| MEK/ERK signaling | KRAS |
| NRAS | |
| BRAF | |
| NF1 | |
| PTPN11 | |
| FGFR3 | |
| NFkB activation | TRAF2 |
| TRAF3 | |
| CYLD | |
| NFKB2 | |
| NFKBIA | |
| BIRC2 | |
| BIRC3 | |
| G1/S cell cycle transition | RB1 |
| CCND1 | |
| CDKN2C | |
| CDKN1B | |
| TP53 | |
| RNA processing | FAM46C |
| DIS3 | |
| Epigenetic regulators | DNMT3A |
| TET2 | |
| KDM6A |
| Favorable | High Risk |
|---|---|
| HRD | del(17p) |
| del(1p32) | |
| t(4;14) | |
| t(14;16) | |
| t(14;20) | |
| gain 1q |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiedmeier-Nutor, J.E.; Bergsagel, P.L. Review of Multiple Myeloma Genetics including Effects on Prognosis, Response to Treatment, and Diagnostic Workup. Life 2022, 12, 812. https://doi.org/10.3390/life12060812
Wiedmeier-Nutor JE, Bergsagel PL. Review of Multiple Myeloma Genetics including Effects on Prognosis, Response to Treatment, and Diagnostic Workup. Life. 2022; 12(6):812. https://doi.org/10.3390/life12060812
Chicago/Turabian StyleWiedmeier-Nutor, Julia Erin, and Peter Leif Bergsagel. 2022. "Review of Multiple Myeloma Genetics including Effects on Prognosis, Response to Treatment, and Diagnostic Workup" Life 12, no. 6: 812. https://doi.org/10.3390/life12060812
APA StyleWiedmeier-Nutor, J. E., & Bergsagel, P. L. (2022). Review of Multiple Myeloma Genetics including Effects on Prognosis, Response to Treatment, and Diagnostic Workup. Life, 12(6), 812. https://doi.org/10.3390/life12060812