
Fetal Brain Development: Regulating Processes and Related Malformations

Abstract
1. Introduction
2. Timetable of Human CNS Development
2.1. Dorsal Induction
2.2. Ventral Induction
2.3. Neurogenesis and Gliogenesis
2.4. Neuronal Migration
2.5. Post-Migration Neuronal Development and Cortical Organization
3. Anomalies of Dorsal Induction-Neural Tube Defects
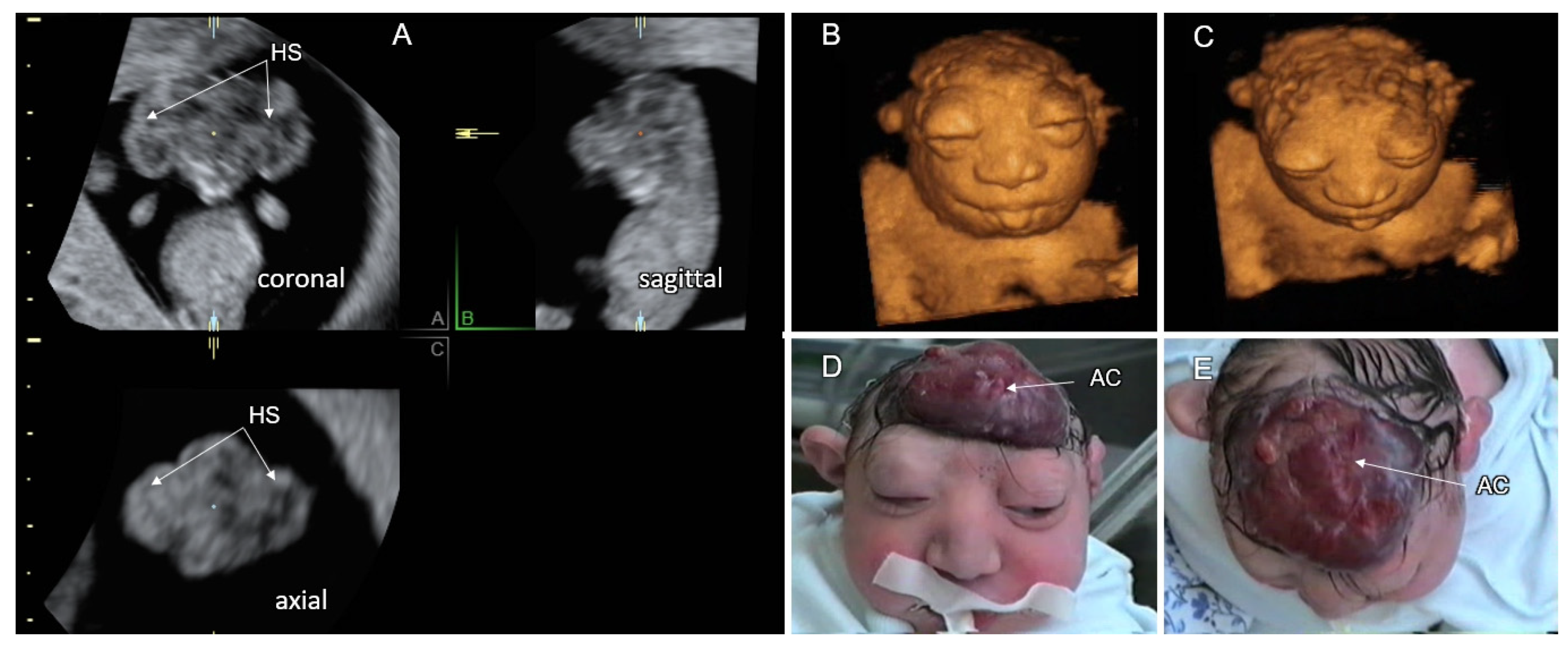
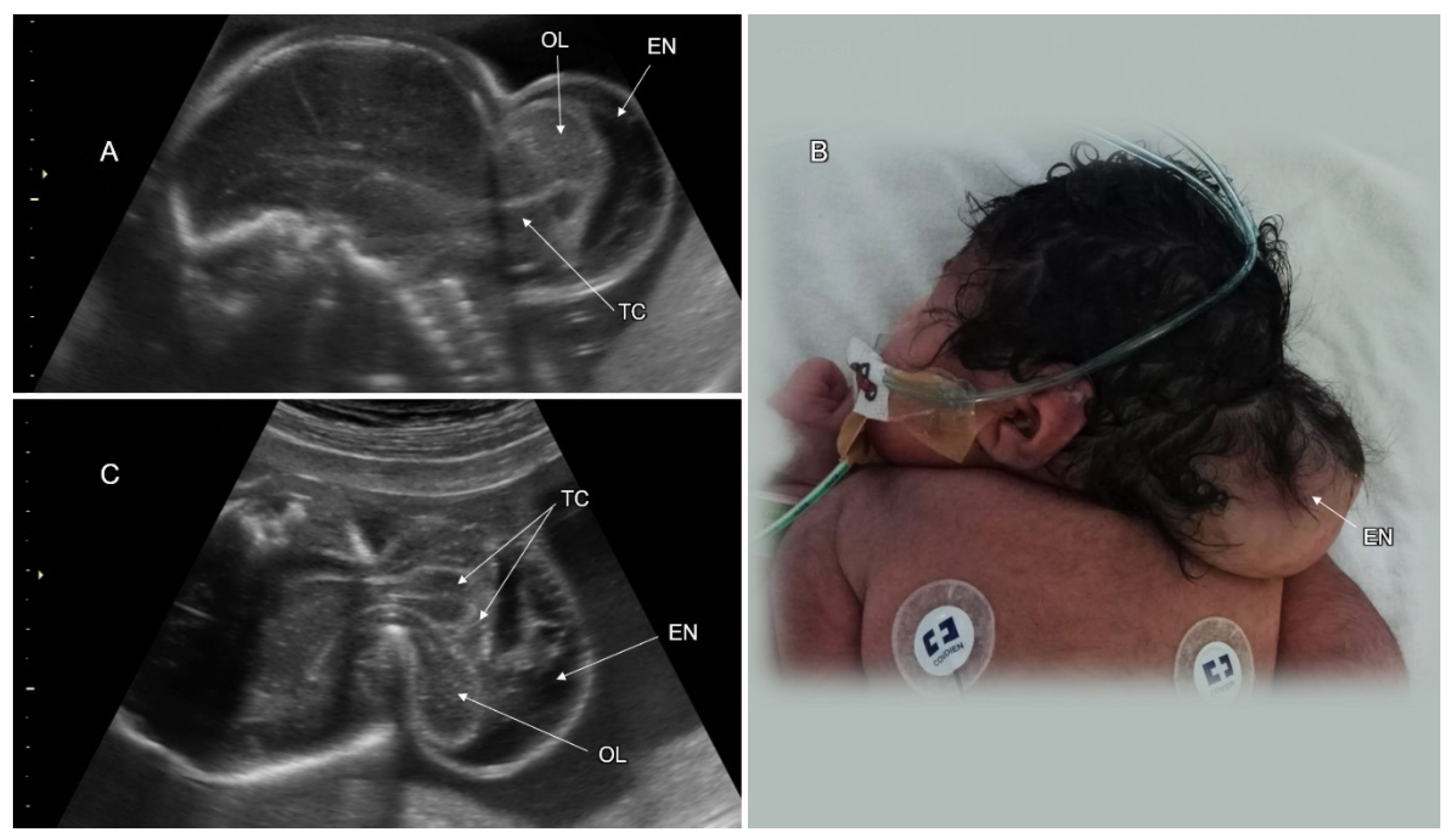
3.1. Cranial NTD
3.2. Spinal Dysraphism
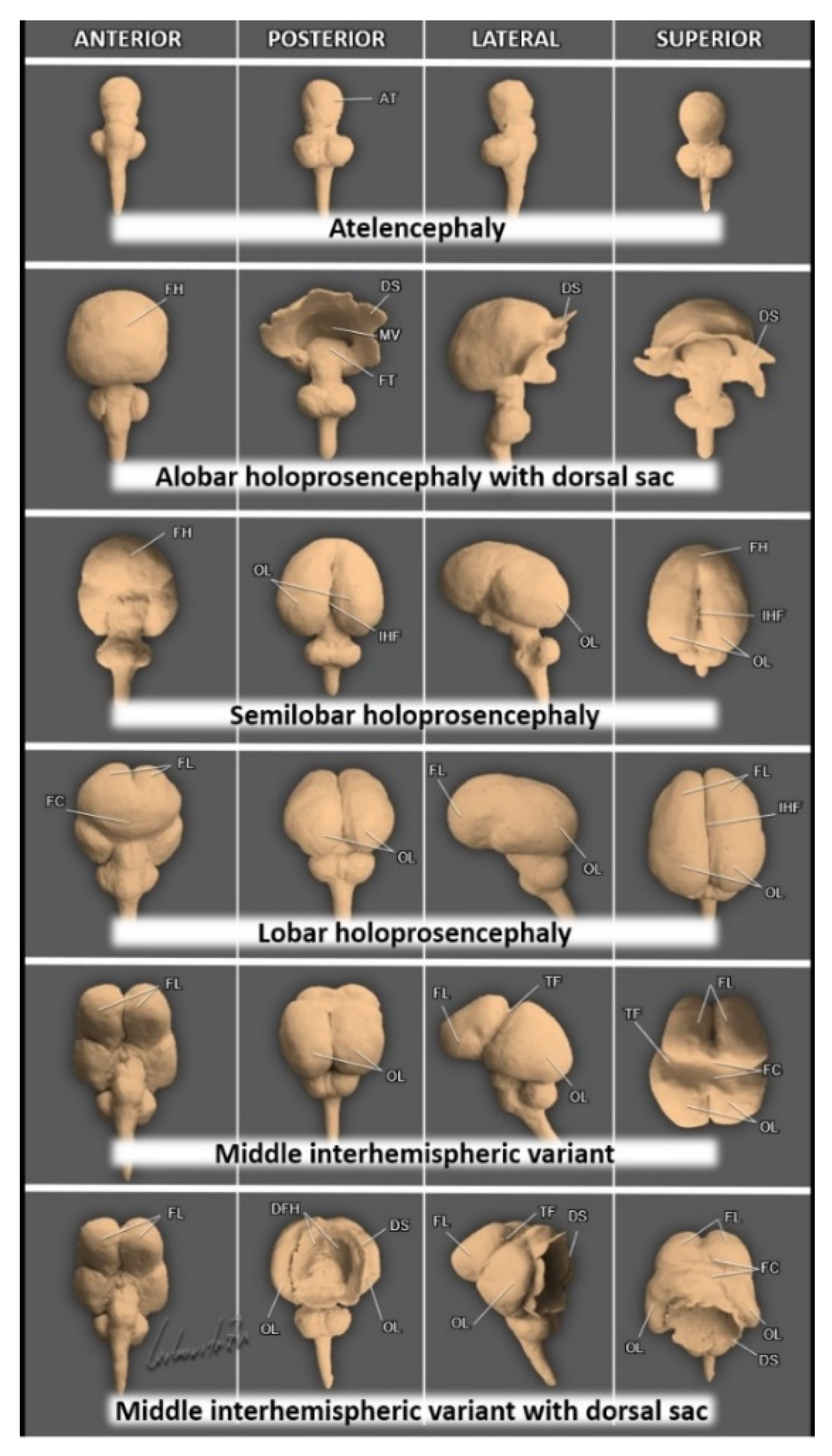
4. Holoprosencephaly—A Disorder of Ventral Induction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Holoprosencephaly | Main Features (See Figure 9) |
|---|---|
| Alobar holoprosencephaly | A complete lack of separation of the cerebral hemispheres (a single-midline ventricle). |
| Semilobar holoprosencephaly | Only anterior lobes fail to separate. |
| Lobar holoprosencephaly | Only the most rostral-inferior parts of the frontal lobes are fused. |
| Middle interhemispheric variant (syntelencephaly) | The posterior frontal and parietal lobes fail to separate. The anterior and occipital cerebral aspects are divided. |
| Septopreoptic holoprosencephaly (minimal form) | A mild subtype of lobar HPE, midline fusion restricted to the septal region or preoptic region of the telencephalon. |
| Microform holoprosencephaly | Only subtle facial midline features: hypotelorism, single maxillary central incisor, and narrowing of the nasal pyriform aperture. |
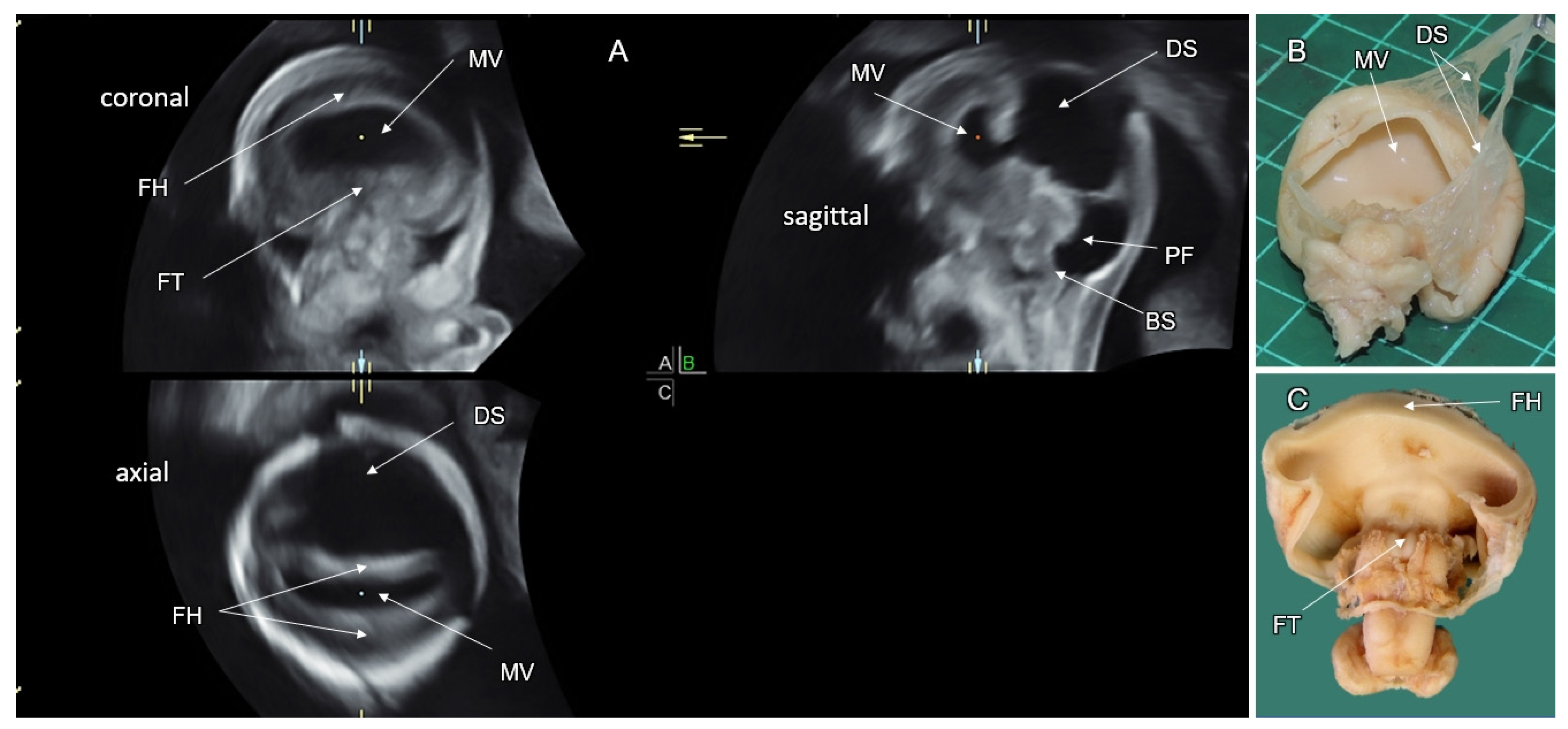
4.1. Alobar HPE
4.2. Semilobar HPE
4.3. Lobar HPE
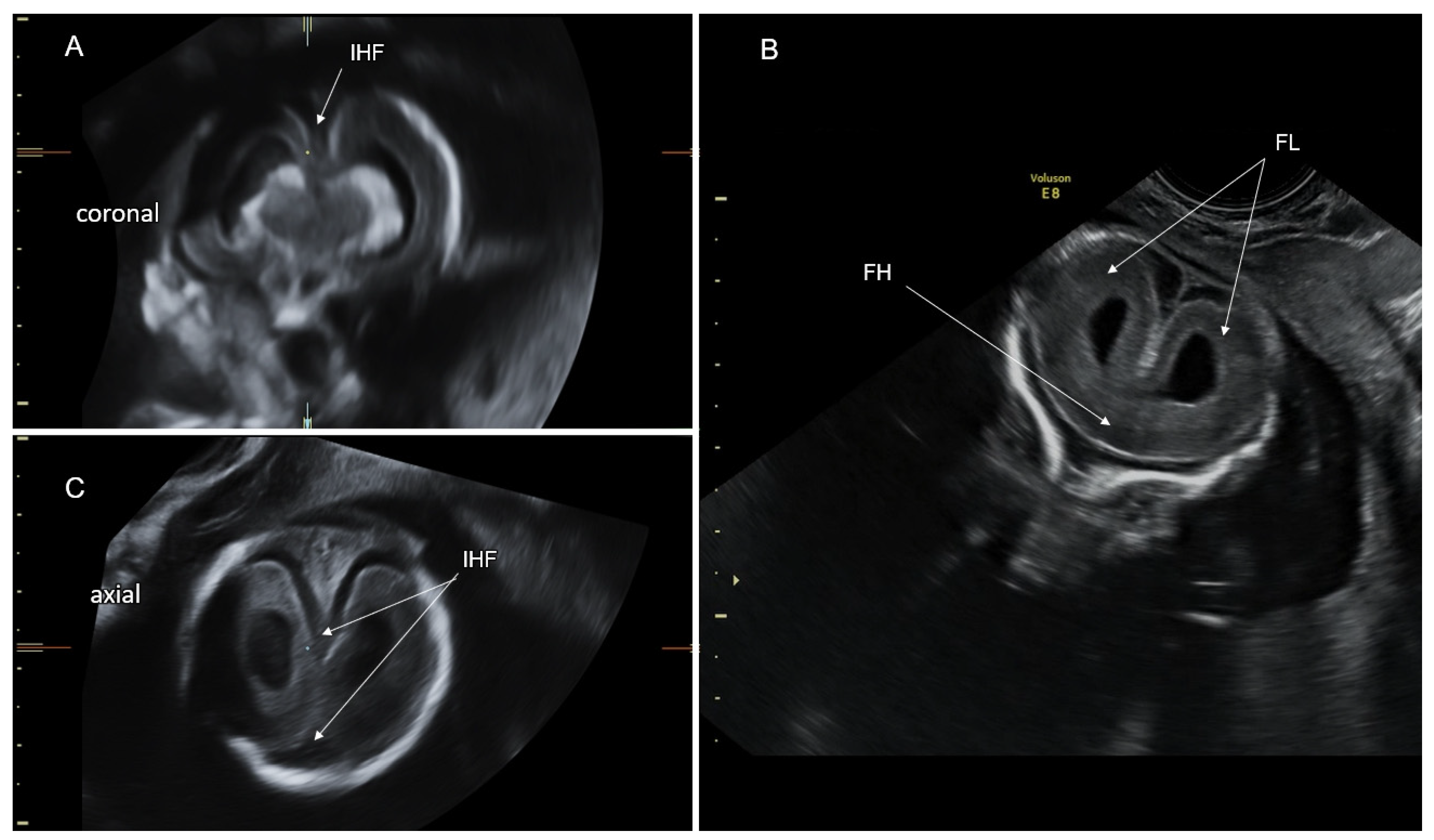
4.4. Middle Interhemispheric Variant
4.5. Milder and Minimal Forms of HPE
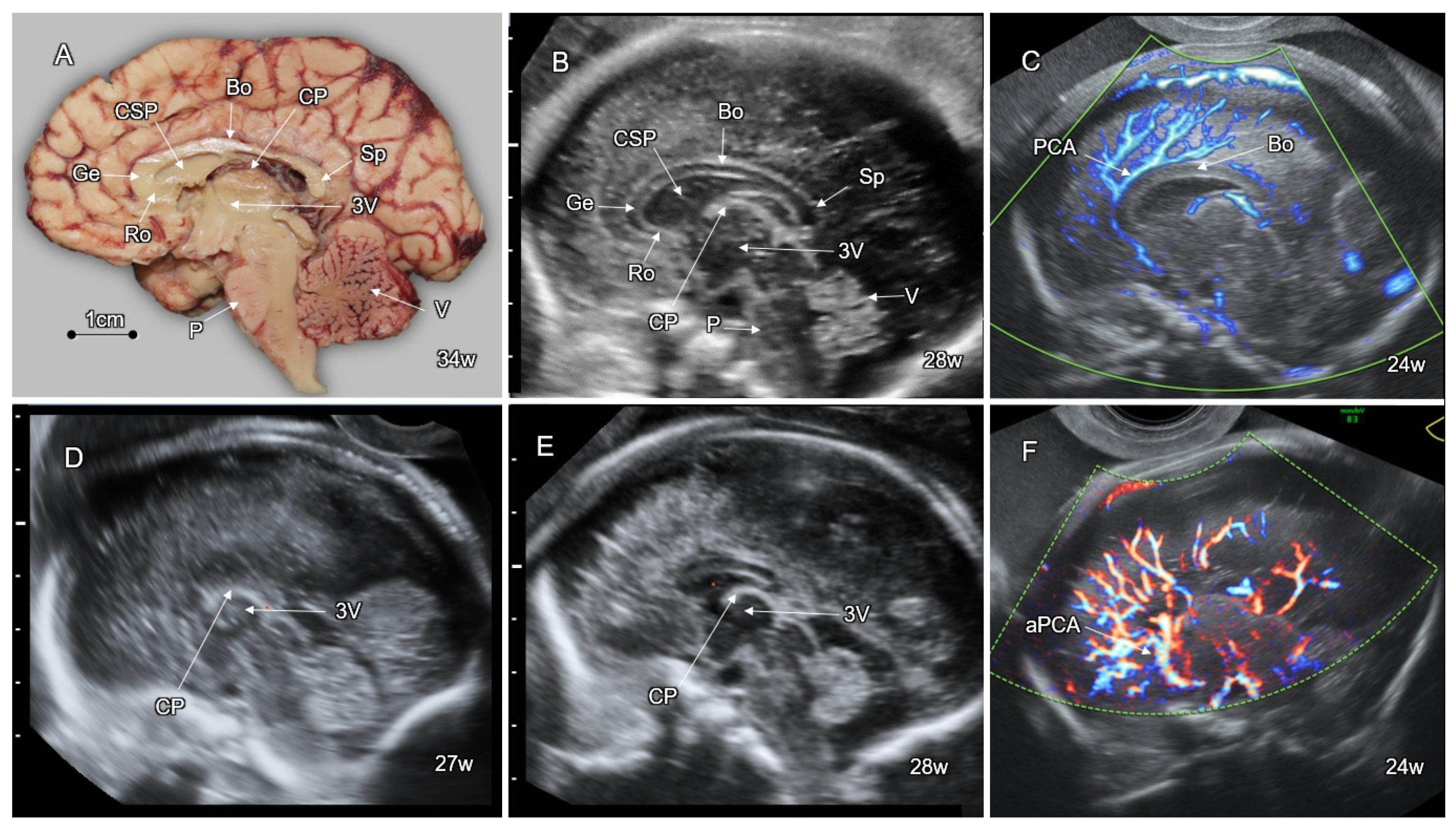
5. Disorders of the Corpus Callosum (DCC)
6. Malformations of Cortical Development
6.1. MCD Secondary to Abnormal Neurogenesis
6.1.1. Microcephaly
6.1.2. Megalencephaly
6.2. MCD Secondary to Abnormal Neuronal Migration
6.2.1. Lissencephaly
6.2.2. Cobblestone Malformation (CM)
6.2.3. Gray Matter Heterotopia
6.3. MCD Due to Abnormal Post-Migration Neuronal Development and Cortical Organization
6.3.1. Polymicrogyria
6.3.2. PMG Associated with Schizencephaly
7. Malformations of the Posterior Fossa
7.1. Dandy–Walker Malformation (DWM)
7.2. Vermian Agenesis (VA), Hypoplasia (VH), and Dysgenesis (VD)
7.3. Blake’s Pouch Cyst (BPC)
7.4. Mega Cisterna Magna (MCM)
7.5. Joubert Syndrome (JS) and JS-Related Disorders
7.6. Rhombencephalosynapsis (RES)
7.7. PF Anomalies Associated with Tubulinopathies
7.8. PF Anomalies Associated with Cobblestone Malformation (CM)
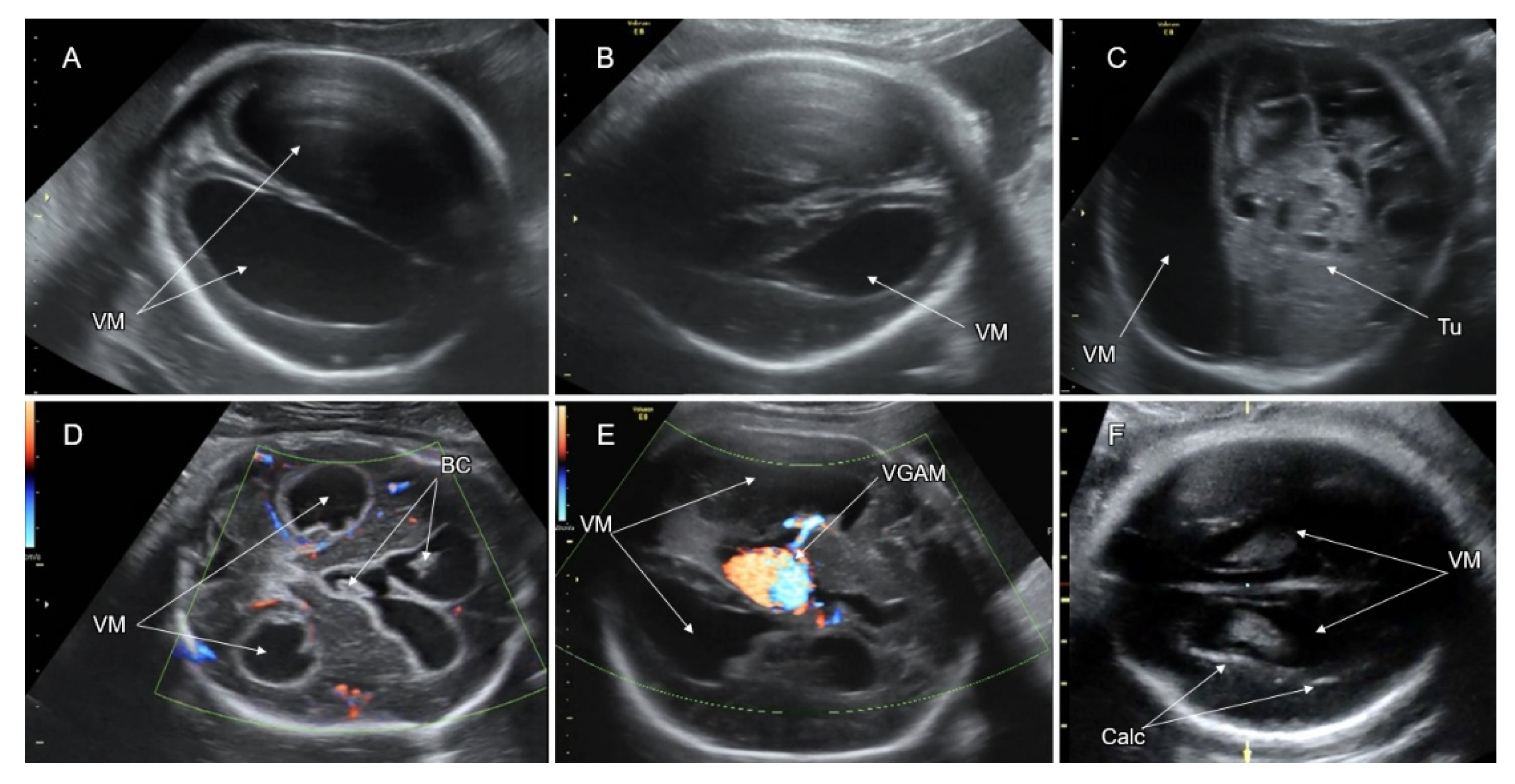
8. Ventriculomegaly
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Paladini, D.; Malinger, G.; Timor-Tritsch, I.E. ISUOG Practice Guidelines (updated), sonographic examination of the fetal central nervous system. Part 2, performance of targeted neurosonography. Ultrasound Obstet. Gynecol. 2021, 57, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Govaert, P.; Triulzi, F. The developing brain by trimester. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 171, pp. 245–289. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, M.I. Multisite neural tube closure in humans. Birth Defects Orig. Artic. Ser. 1996, 30, 203–225. [Google Scholar] [PubMed]
- ten Donkelaar, H.; van der Vliet, T. Overview of the Development of the Human Brain and Spinal Cord. In Clinical Neuroembryology, 1st ed.; ten Donkelaar, H., Lammens, M., Eds.; Springer: Würzburg, Germany, 2006; pp. 1–40. [Google Scholar]
- Rowitch, D.H.; Kriegstein, A.R. Developmental genetics of vertebrate glial-cell specification. Nature 2010, 468, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Guérout, N.; Li, X. Cell fate control in the developing central nervous system. Exp. Cell Res. 2014, 321, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Obernier, K. Adult neural stem cells bridge their niche. Cell Stem Cell 2012, 10, 698–708. [Google Scholar] [CrossRef]
- Bystron, I.; Blakemore, C. Development of the human cerebral cortex, Boulder Committee revisited. Nat. Rev. Neurosci. 2008, 9, 110–122. [Google Scholar] [CrossRef]
- Taverna, E.; Götz, M. The cell biology of neurogenesis, toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol. 2014, 30, 465–502. [Google Scholar] [CrossRef]
- Linderkamp, O.; Janus, L. Time Table of Normal Foetal Brain Development. Int. J. Prenat. Perinat. Psychol. Med. 2009, 21, 4–16. [Google Scholar]
- O’Rahilly, R.; Muller, F. The Embryonic Human Brain Atlas of Developmental Stages, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006; pp. 31–38. [Google Scholar]
- Saxena, S.; Caroni, P. Mechanisms of axon degeneration, from development to disease. Prog. Neurobiol. 2007, 83, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.K.; Woods, C.G. Primary microcephaly, do all roads lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S.; Cai, X. Human mutations in NDE1 cause extreme microcephaly with lissencephaly. Am. J. Hum. Genet. 2011, 88, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Gilmore, E.C. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 2010, 42, 245–249. [Google Scholar] [CrossRef]
- Griffith, E.; Walker, S. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008, 40, 232–236. [Google Scholar] [CrossRef]
- Desir, J.; Cassart, M. Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero-posterior gradient pre- and post-natally. Am. J. Med. Genet. A 2008, 146, 1439–1443. [Google Scholar] [CrossRef]
- Kumar, A.; Girimaji, S.C. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet. 2009, 84, 286–290. [Google Scholar] [CrossRef]
- Yu, T.W.; Mochida, G.H. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef]
- Feng, Y.; Walsh, C.A. Mitotic spindle regulation by Nde1 controls cerebral cortical size. J. Neuron 2004, 44, 279–293. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Guerrini, R. A developmental and genetic classification for malformations of cortical development, update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef]
- Myshrall, T.D.; Moore, S.A. Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex. J. Neuropathol. Exp. Neurol. 2012, 71, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S. Molecular genetics of neuronal migration disorders. Curr. Neurol. Neurosci. Rep. 2011, 11, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, A.L.; Faust, P.L. New directions for neuronal migration. Curr. Opin. Neurobiol. 1998, 8, 45–54. [Google Scholar] [CrossRef]
- Marín, O.; Yaron, A. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science 2001, 293, 872–875. [Google Scholar] [CrossRef]
- Anderson, S.A.; Marín, O. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development 2001, 128, 353–363. [Google Scholar] [CrossRef]
- Trinidad, M.C.; Wick, M. ACOG PRACTICE BULLETIN No. 187, Neural Tube Defects. Obstet. Gynecol. 2017, 130, E279–E290. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Kibar, Z. Genetic basis of neural tube defects. Semin. Pediatr. Neurol. 2009, 16, 101–110. [Google Scholar] [CrossRef]
- Chatzipapas, I.K.; Whitlow, B.J. The “Mickey Mouse” sign and the diagnosis of anencephaly in early pregnancy. Ultrasound Obstet. Gynecol. 1999, 13, 196–199. [Google Scholar] [CrossRef]
- Johnson, S.P.; Sebire, N.J. Ultrasound screening for anencephaly at 10–14 weeks of gestation. Ultrasound Obstet. Gynecol. 1997, 9, 14–16. [Google Scholar] [CrossRef]
- Lo, B.W.Y.; Kulkarni, A.V. Clinical predictors of developmental outcome in patients with cephaloceles. J. Neurosurg. Pediatr. 2008, 2, 254–257. [Google Scholar] [CrossRef]
- Favoreel, N.; Devooghdt, M. Atretic cephalocele. Jbr-Btr 2015, 98, 119–120. [Google Scholar] [CrossRef]
- Monteagudo, A. Posterior Encephalocele. Am. J. Obstet. Gynecol. 2020, 223, B9–B12. [Google Scholar] [CrossRef] [PubMed]
- Paladini, D.; Volpe, P. Ultrasound of Congenital Fetal Anomalies Differential Diagnosis and Prognostic Indicators, 1st ed.; Informa: London, UK, 2007; pp. 52–62. [Google Scholar]
- Martínez-Lage, J.F.; Poza, M. The child with a cephalocele, etiology.; neuroimaging.; and outcome. Childs Nerv. Syst. 1996, 12, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kumar, A. Congenital spinal cord anomalies, a pictorial review. Curr. Probl. Diagn. Radiol. 2013, 42, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rufener, S.L.; Ibrahim, M. Congenital spine and spinal cord malformations—Pictorial review. Am. J. Roentgenol. 2010, 194, S26–S37. [Google Scholar] [CrossRef] [PubMed]
- Masini, L.; De Luca, C. Prenatal diagnosis, natural history, postnatal treatment and outcome of 222 cases of spina bifida: Experience of a tertiary center. Ultrasound Obstet. Gynecol. 2019, 53, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, K.H.; Gabbe, S.G. Ultrasound Screening for Spina Bifida, Cranial and Cerebellar Signs. Lancet 1986, 328, 72–74. [Google Scholar] [CrossRef]
- Van den Hof, M.C.; Nicolaides, K.H. Evaluation of the lemon and banana signs in one hundred thirty fetuses with open spina bifida. Am. J. Obstet. Gynecol. 1990, 162, 322–327. [Google Scholar] [CrossRef]
- Chaoui, R.; Benoit, B. Assessment of intracranial translucency (IT) in the detection of spina bifida at the 11–13-week scan. Ultrasound Obstet. Gynecol. 2009, 34, 249–252. [Google Scholar] [CrossRef]
- Fong, K.W.; Toi, A. Retrospective review of diagnostic performance of intracranial translucency in detection of open spina bifida at the 11–13-week scan. Ultrasound Obstet. Gynecol. 2011, 38, 630–634. [Google Scholar] [CrossRef]
- Sepulveda, W.; Corral, E. Chromosomal abnormalities in fetuses with open neural tube defects, prenatal identification with ultrasound. Ultrasound Obstet. Gynecol. 2004, 23, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Hume, R.F., Jr.; Drugan, A.; Reichler, A.; Lampinen, J.; Martin, L.S.; Johnson, M.P.; Evans, M.I. Aneuploidy among prenatally detected neural tube defects. Am. J. Med. Genet. 1996, 61, 171–173. [Google Scholar] [CrossRef]
- Santirocco, M.; Plaja, A. Chromosomal microarray analysis in fetuses with central nervous system anomalies: An 8-year long observational study from a tertiary care university hospital. Prenat. Diagn. 2021, 41, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P. Syndromes, disorders and maternal risk factors associated with neural tube defects (I). Taiwan J. Obstet. Gynecol. 2008, 47, 1–9. [Google Scholar] [CrossRef]
- Chen, C.P. Syndromes, disorders and maternal risk factors associated with neural tube defects (II). Taiwan J. Obstet. Gynecol. 2008, 47, 10–17. [Google Scholar] [CrossRef]
- MRC Vitamin Study Research Group. Prevention of neural tube defects: Results of the Medical Research Council Vitamin study. Obstet. Gynecol. Surv. 1992, 47, 21–23. [Google Scholar] [CrossRef]
- Mohd-Zin, S.W.; Marwan, A.I. Spina bifida: Pathogenesis, mechanisms and genes in mice and humans. Scientifica 2017, 2017, 1–29. [Google Scholar] [CrossRef]
- Greene, N.D.E.; Stanier, P. Genetics of human neural tube defects. Hum. Mol. Genet. 2009, 18, R113–R129. [Google Scholar] [CrossRef]
- Adzick, N.S.; Sutton, L.N. Successful fetal surgery for spina bifida. Lancet 1998, 352, 1675–1676. [Google Scholar] [CrossRef]
- Inversetti, A.; Van der Veeken, L. Neurodevelopmental outcome of children with spina bifida aperta repaired prenatally vs. postnatally, systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2019, 53, 293–301. [Google Scholar] [CrossRef]
- Johnson, C.Y.; Honein, M.A. Pregnancy termination following prenatal diagnosis of anencephaly or spina bifida, a systematic review of the literature. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Trudell, A.S.; Odibo, A.O. Diagnosis of spina bifida on ultrasound, always termination? Best Pract. Res. Clin. Obstet. Gynaecol. 2014, 28, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Volpe, P.; Campobasso, G. Disorders of prosencephalic development. Prenat. Diagn. 2009, 29, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.P.; Townsend, J.J. Atelencephalic aprosencephaly. J. Child. Neurol. 1994, 9, 412–416. [Google Scholar] [CrossRef]
- Winter, T.C.; Kennedy, A.M. Holoprosencephaly: A survey of the entity, with embryology and fetal imaging. Radiographics 2015, 35, 275–290. [Google Scholar] [CrossRef]
- DeMyer, W.; Zeman, W.P.C. The face predicts the brain: Diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics 1964, 34, 256–263. [Google Scholar] [CrossRef]
- Marcorelles, P.; Laquerriere, A. Neuropathology of holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 109–119. [Google Scholar] [CrossRef]
- Pilu, G.; Ambrosetto, P.; Ancora, G. Intraventricular fused fornices: A specific sign of fetal lobar holoprosencephaly. Ultrasound Obstet. Gynecol. 1994, 4, 65–67. [Google Scholar] [CrossRef]
- Bernard, J.P.; Drummond, C.L. A new clue to the prenatal diagnosis of lobar holoprosencephaly: The abnormal pathway of the anterior cerebral artery crawling under the skull. Ultrasound Obstet. Gynecol. 2002, 19, 605–607. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Quint, D.J. Middle interhemispheric fusion: An unusual variant of holoprosencephaly. AJNR Am. J. Neuroradiol. 1993, 14, 431–440. [Google Scholar]
- Lewis, A.J.; Simon, E.M. Middle interhemispheric variant of holoprosencephaly: A distinct cliniconeuroradiologic subtype. Neurology 2002, 59, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S.; Barnes, P.D. Neuroimaging advances in holoprosencephaly: Refining the spectrum of the midline malformation. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, W.; Dezerega, V. First-trimester sonographic diagnosis of holoprosencephaly: Value of the “butterfly” sign. J. Ultrasound Med. 2004, 23, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Levey, E.B.; Stashinko, E. Management of children with holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 183–190. [Google Scholar] [CrossRef]
- Kauvar, E.F.; Muenke, M. Holoprosencephaly: Recommendations for diagnosis and management. Curr. Opin. Pediatr. 2010, 22, 687–695. [Google Scholar] [CrossRef]
- Paul, L.K.; Brown, W.S. Agenesis of the corpus callosum: Genetics, developmental and functional aspects of connectivity. Nat. Rev. Neurosci. 2007, 8, 287–299. [Google Scholar] [CrossRef]
- Edwards, T.J.; Sherr, E.H. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014, 137, 1579–1613. [Google Scholar] [CrossRef]
- Wong, B.K.Y.; Sutton, V.R. Aicardi syndrome, an unsolved mystery: Review of diagnostic features, previous attempts, and future opportunities for genetic examination. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 423–431. [Google Scholar] [CrossRef]
- Birnbaum, R.; Barzilay, R. The early pattern of human corpus callosum development: A transvaginal 3D neurosonographic study. Prenat. Diagn. 2020, 40, 1239–1245. [Google Scholar] [CrossRef]
- Leombroni, M.; Khalil, A. Fetal midline anomalies: Diagnosis and counselling Part 1: Corpus callosum anomalies. Eur. J. Paediatr. Neurol. 2018, 22, 951–962. [Google Scholar] [CrossRef]
- Diogo, M.C.; Glatter, S. Improved neurodevelopmental prognostication in isolated corpus callosal agenesis, fetal magnetic resonance imaging-based scoring system. Ultrasound Obstet. Gynecol. 2021, 58, 34–41. [Google Scholar] [CrossRef] [PubMed]
- des Portes, V.; Rolland, A. Outcome of isolated agenesis of the corpus callosum: A population-based prospective study. Eur. J. Paediatr. Neurol. 2018, 22, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Putoux, A.; Nampoothiri, S. Novel KIF7 mutations extend the phenotypic spectrum of acrocallosal syndrome. J. Med. Genet. 2012, 49, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.D. Smith–Lemli–Opitz syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2008, 16, 535–541. [Google Scholar] [CrossRef]
- Lia, N.; Sama, S. FOXG1 syndrome: A rare disorder often misdiagnosed as cerebral palsy. Neurology 2019, 92, 15. [Google Scholar]
- Krupa, K.; Bekiesinska-Figatowska, M. Congenital and acquired abnormalities of the corpus callosum: A pictorial essay. Biomed. Res. Int. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Atallah, A.; Lacalm, A. Prenatal diagnosis of pericallosal curvilinear lipoma: Specific imaging pattern and diagnostic pitfalls. Ultrasound Obstet. Gynecol. 2018, 51, 269–273. [Google Scholar] [CrossRef]
- Restrepo, L.R.; Camacho López, P.A. Diagnostic approach to the alterations of the corpus callosum: State of the art. Rev. Colomb. Radiol. 2019, 30, 5147–5152. [Google Scholar]
- Khalil, A.; Sotiriadis, A. ISUOG Practice Guidelines: Role of ultrasound in congenital infection. Ultrasound Obstet. Gynecol. 2020, 56, 128–151. [Google Scholar] [CrossRef]
- Malinger, G.; Paladini, D. ISUOG Practice Guidelines (updated): Sonographic examination of the fetal central nervous system. Part 1: Performance of screening examination and indications for targeted neurosonography. Ultrasound Obstet. Gynecol. 2020, 56, 476–484. [Google Scholar] [CrossRef]
- Karl, K.; Esser, T. Cavum septi pellucidi (CSP) ratio: A marker for partial agenesis of the fetal corpus callosum. Ultrasound Obstet. Gynecol. 2017, 50, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Shen, O.; Gelot, A.B. Abnormal shape of the cavum septi pellucidi: An indirect sign of partial agenesis of the corpus callosum. Ultrasound Obstet. Gynecol. 2015, 46, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.H.; Bartha, A.I. Agenesis of the corpus callosum: An MR imaging analysis of associated abnormalities in the fetus. AJNR Am. J. Neuroradiol. 2009, 30, 257. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.N.; Malinger, G.T. Primary disorders of metabolism and disturbed fetal brain development. Clin. Perinatol. 2009, 36, 621–638. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, F.; Pagani, G. Outcomes associated with isolated agenesis of the corpus callosum: A meta-analysis. Pediatrics 2016, 138, e20160445. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.; Llinares-Benadero, C. Cerebral cortex expansion and folding: What have we learned? EMBO J. 2016, 35, 1021–1044. [Google Scholar] [CrossRef]
- Desikan, R.S.; Barkovich, A.J. Malformations of cortical development. Ann. Neurol. 2016, 80, 797–810. [Google Scholar] [CrossRef]
- Subramanian, L.; Calcagnotto, M.E. Cortical malformations: Lessons in human brain development. Front. Cell Neurosci. 2020, 13, 576. [Google Scholar] [CrossRef]
- Guerrini, R.; Dobyns, W.B. Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 2014, 13, 710. [Google Scholar] [CrossRef]
- Gilmore, E.C.; Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 461. [Google Scholar] [CrossRef]
- Leibovitz, Z.; Lerman-Sagie, T. Diagnostic approach to fetal microcephaly. Eur. J. Paediatr. Neurol. 2018, 22, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Ashwal, S.; Michelson, D. Quality standards subcommittee of the American Academy of Neurology and the practice committee of the Child Neurology Society. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review), report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2009, 73, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Chervenak, F.A.; Jeanty, P. The diagnosis of fetal microcephaly. Am. J. Obstet. Gynecol. 1984, 149, 512–517. [Google Scholar] [CrossRef]
- Society for Maternal-Fetal Medicine Publications Committee. Ultrasound screening for fetal microcephaly following Zika virus exposure. Am. J. Obstet. Gynecol. 2016, 214, B2–B4. [Google Scholar] [CrossRef]
- Leibovitz, Z.; Daniel-Spiegel, E. Prediction of microcephaly at birth using three reference ranges for fetal head circumference: Can we improve prenatal diagnosis? Ultrasound Obstet. Gynecol. 2016, 47, 586–592. [Google Scholar] [CrossRef]
- Leibovitz, Z.; Shiran, C. Application of a novel prenatal vertical cranial biometric measurement can improve accuracy of microcephaly diagnosis in utero. Ultrasound Obstet. Gynecol. 2016, 47, 593–599. [Google Scholar] [CrossRef]
- Dolk, H. The Predictive Value of Microcephaly During the First Year of Life for Mental Retardation At Seven Years. Dev. Med. Child Neurol. 1991, 33, 974–983. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Rios, J.J. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis and evaluation. Am. J. Med. Genet. Part A 2015, 167, 287–295. [Google Scholar] [CrossRef]
- Pavone, P.; Praticò, A.D. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine 2017, 96, e6814. [Google Scholar] [CrossRef]
- Severino, M.; Geraldo, A.F. Definitions and classification of malformations of cortical development, practical guidelines. Brain 2020, 143, 2874–2894. [Google Scholar] [CrossRef]
- Tan, A.P.; Chong, W.K. Comprehensive genotype-phenotype correlation in lissencephaly. Quant. Imag. Med. Surg. 2018, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Dobyns, W.B. Lissencephaly and the molecular basis of neuronal migration. Hum. Mol. Genet. 2003, 12, R89–R96. [Google Scholar] [CrossRef] [PubMed]
- Herman, T.E.; Siegel, M.J. Miller-Dieker syndrome, type 1 lissencephaly. J. Perinatol. 2008, 28, 313–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pilz, D.T.; Quarrell, W.J. Syndromes with lissencephaly. J. Med. Genet. 1996, 33, 319–323. [Google Scholar] [CrossRef]
- Lerman-Sagie, T.; Pogledic, I. A practical approach to prenatal diagnosis of malformations of cortical development. Eur. J. Paediatr. Neurol. 2021, 34, 50–61. [Google Scholar] [CrossRef]
- Squier, W.; Jansen, A. Polymicrogyria: Pathology, fetal origins and mechanisms. Acta Neuropathol. Commun. 2014, 2, 1–16. [Google Scholar] [CrossRef]
- Leventer, R.J.; Jansen, A. Clinical and imaging heterogeneity of polymicrogyria: A study of 328 patients. Brain 2010, 133, 1415–1427. [Google Scholar] [CrossRef]
- Pang, T.; Atefy, R. Malformations of cortical development. Neurologist 2008, 14, 181–191. [Google Scholar] [CrossRef]
- Barkovich, J.A. Developmental disorders of the midbrain and hindbrain. Front. Neuroanat. 2012, 6, 7. [Google Scholar] [CrossRef]
- Severino, M.; Huisman, T.A.G.M. Posterior Fossa Malformations. Neuroimaging Clin. N. Am. 2019, 29, 367–383. [Google Scholar] [CrossRef]
- Garel, C.; Fallet-Bianco, C. The fetal cerebellum: Development and common malformations. J. Child Neurol. 2011, 26, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Guibaud, L. Practical approach to prenatal posterior fossa abnormalities using MRI. Pediatr. Radiol. 2004, 34, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Garel, C. Posterior fossa malformations, main features and limits in prenatal diagnosis. Pediatr. Radiol. 2010, 40, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Malinger, G.; Lev, D. The fetal cerebellum. Pitfalls in diagnosis and management. Prenat. Diagn. 2009, 29, 372–380. [Google Scholar] [CrossRef]
- Leibovitz, Z.; Shkolnik, C. Assessment of fetal midbrain and hindbrain in mid-sagittal cranial plane by three-dimensional multiplanar sonography. Part 2: Application of nomograms to fetuses with posterior fossa malformations. Ultrasound Obstet. Gynecol. 2014, 44, 581–587. [Google Scholar] [CrossRef]
- D’Antonio, F.; Khalil, A. Systematic review and meta-analysis of isolated posterior fossa malformations on prenatal ultrasound imaging (part 1): Nomenclature, diagnostic accuracy and associated anomalies. Ultrasound Obstet. Gynecol. 2016, 47, 690–697. [Google Scholar] [CrossRef]
- D’Antonio, F.; Khalil, A. Systematic review and meta-analysis of isolated posterior fossa malformations on prenatal imaging (part 2): Neurodevelopmental outcome. Ultrasound Obstet. Gynecol. 2016, 48, 28–37. [Google Scholar] [CrossRef]
- Volpe, P.; Contro, E. Brainstem–vermis and brainstem–tentorium angles allow accurate categorization of fetal upward rotation of cerebellar vermis. Ultrasound Obstet. Gynecol. 2012, 39, 632–635. [Google Scholar] [CrossRef]
- Haratz, K.K.; Shulevitz, S.L. Fourth ventricle index: Sonographic marker for severe fetal vermian dysgenesis/agenesis. Ultrasound Obstet. Gynecol. 2019, 53, 390–395. [Google Scholar] [CrossRef]
- Krajden Haratz, K.; Oliveira Szejnfeld, P. Prenatal diagnosis of rhombencephalosynapsis: Neuroimaging features and severity of vermian anomaly. Ultrasound Obstet. Gynecol. 2021, 58, 864–874. [Google Scholar] [CrossRef]
- Almog, B.; Gamzu, R. Fetal lateral ventricular width: What should be its upper limit? A prospective cohort study and reanalysis of the current and previous data. J. Ultrasound Med. 2003, 22, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Guibaud, L. Fetal cerebral ventricular measurement and ventriculomegaly, time for procedure standardization. Ultrasound Obstet. Gynecol. 2009, 34, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.S.; Monteagudo, A. Mild fetal ventriculomegaly: Diagnosis, evaluation and management. Am. J. Obstet. Gynecol. 2018, 219, B2–B9. [Google Scholar] [CrossRef] [PubMed]
- Salomon, L.J.; Bernard, J.P. Reference ranges for fetal ventricular width: A non-normal approach. Ultrasound Obstet. Gynecol. 2007, 30, 61–66. [Google Scholar] [CrossRef]
- Gaglioti, P.; Oberto, M. The significance of fetal ventriculomegaly: Etiology, short-and long-term outcomes. Prenat. Diagn. 2009, 29, 381–388. [Google Scholar] [CrossRef]
- Pagani, G.; Thilaganathan, B. Neurodevelopmental outcome in isolated mild fetal ventriculomegaly, systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2014, 44, 254–260. [Google Scholar] [CrossRef]
- Katorza, E.; Duvdevani, N. Coronal approach for measuring both fetal lateral ventricles: Is there an advantage over the axial view? Prenat. Diagn. 2014, 34, 279–284. [Google Scholar] [CrossRef]
- Melchiorre, K.; Bhide, A. Counseling in isolated mild fetal ventriculomegaly. Ultrasound Obstet. Gynecol. 2009, 34, 212–224. [Google Scholar] [CrossRef]
- Guibaud, L.; Lacalm, A. Etiological diagnostic tools to elucidate ‘isolated’ ventriculomegaly. Ultrasound Obstet. Gynecol. 2015, 46, 1–11. [Google Scholar] [CrossRef]
- Mohammad, K.; Scott, J.N. Consensus approach for standardizing the screening and classification of preterm brain injury diagnosed with cranial ultrasound: A Canadian perspective. Front. Pediatr. 2021, 9, 618236. [Google Scholar] [CrossRef]
- Li, Z.; Fu, F. Application of chromosome microarray analysis for the delineation of pathogenesis for fetal ventriculomegaly. Chin. J. Med. Genet. 2017, 34, 576–582. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, Y. Chromosomal microarray analysis for lateral ventriculomegaly in fetus. Chin. J. Med. Genet. 2015, 32, 789–792. [Google Scholar] [CrossRef]
















| Developmental Process | Gestational Age (Postmenstrual Weeks) | Main Features | Related Anomalies |
|---|---|---|---|
| Dorsal induction | 5–7 | Formation of the neural tube. | Neural tube defects (anencephaly, cephalocele, spina bifida). |
| Ventral induction | 6–9 | Division of the prosencephalon into two separate telencephalic vesicles (future cerebral hemispheres), formation of optic vesicles, olfactory bulbs, and corresponding facial structures. | Holoprosencephaly. |
| Neuronal/glial proliferation | Beginning at the 10th week, maximal rate at 17–18 weeks, ending at the late 2nd trimester. | Increase in population of CNS cell. The excessive cells undergo apoptosis. | Microcephaly, megalencephaly, hemimegalencephaly. |
| Neuronal migration | 12–20 | Movement of neural cells from the subventricular zone towards the outer zones of the developing brain, cortical formation. | Lissencephaly, cobblestone malformation, gray matter heterotopia. |
| Post-migration neuronal development and cortical organization | From 22 weeks to postnatal period. | Cortical maturation, outgrowth of axons and dendrites from cortical neurons, and synaptogenesis. | Polymicrogyria, cortical dysplasia. |
| Type of the NTD * | Risk of Chromosomal Anomalies | Associated Anomalies/Syndromes | Outcome |
|---|---|---|---|
| Anencephaly | Low |
| Incompatible with life |
| Cephalocele | 14–18% |
| Childhood mortality:
|
| Spinal dysraphism | 2–16% |
| OSD # outcomes:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leibovitz, Z.; Lerman-Sagie, T.; Haddad, L. Fetal Brain Development: Regulating Processes and Related Malformations. Life 2022, 12, 809. https://doi.org/10.3390/life12060809
Leibovitz Z, Lerman-Sagie T, Haddad L. Fetal Brain Development: Regulating Processes and Related Malformations. Life. 2022; 12(6):809. https://doi.org/10.3390/life12060809
Chicago/Turabian StyleLeibovitz, Zvi, Tally Lerman-Sagie, and Leila Haddad. 2022. "Fetal Brain Development: Regulating Processes and Related Malformations" Life 12, no. 6: 809. https://doi.org/10.3390/life12060809
APA StyleLeibovitz, Z., Lerman-Sagie, T., & Haddad, L. (2022). Fetal Brain Development: Regulating Processes and Related Malformations. Life, 12(6), 809. https://doi.org/10.3390/life12060809