
Metal Ion-Directed Specific DNA Structures and Their Functions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
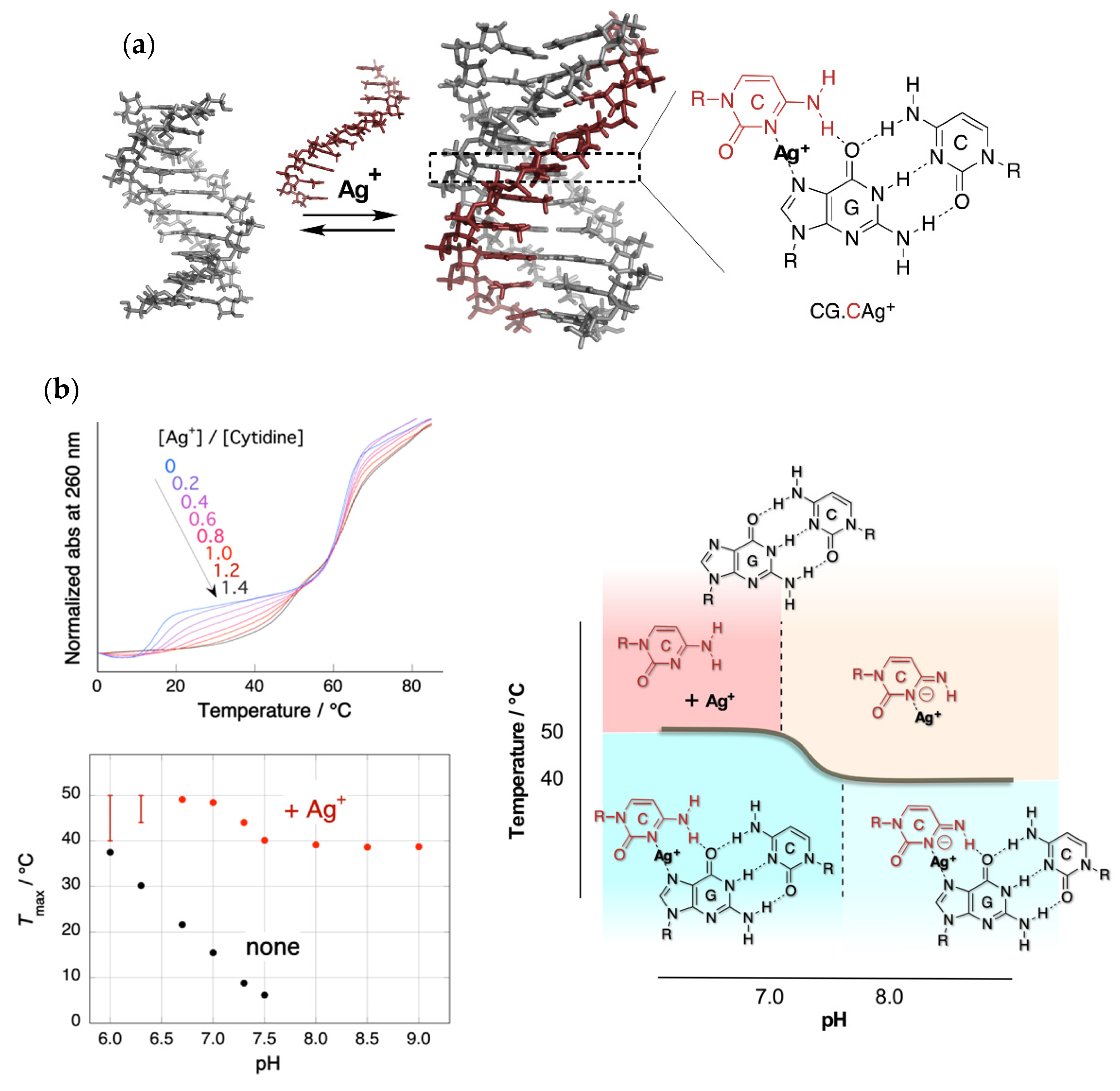
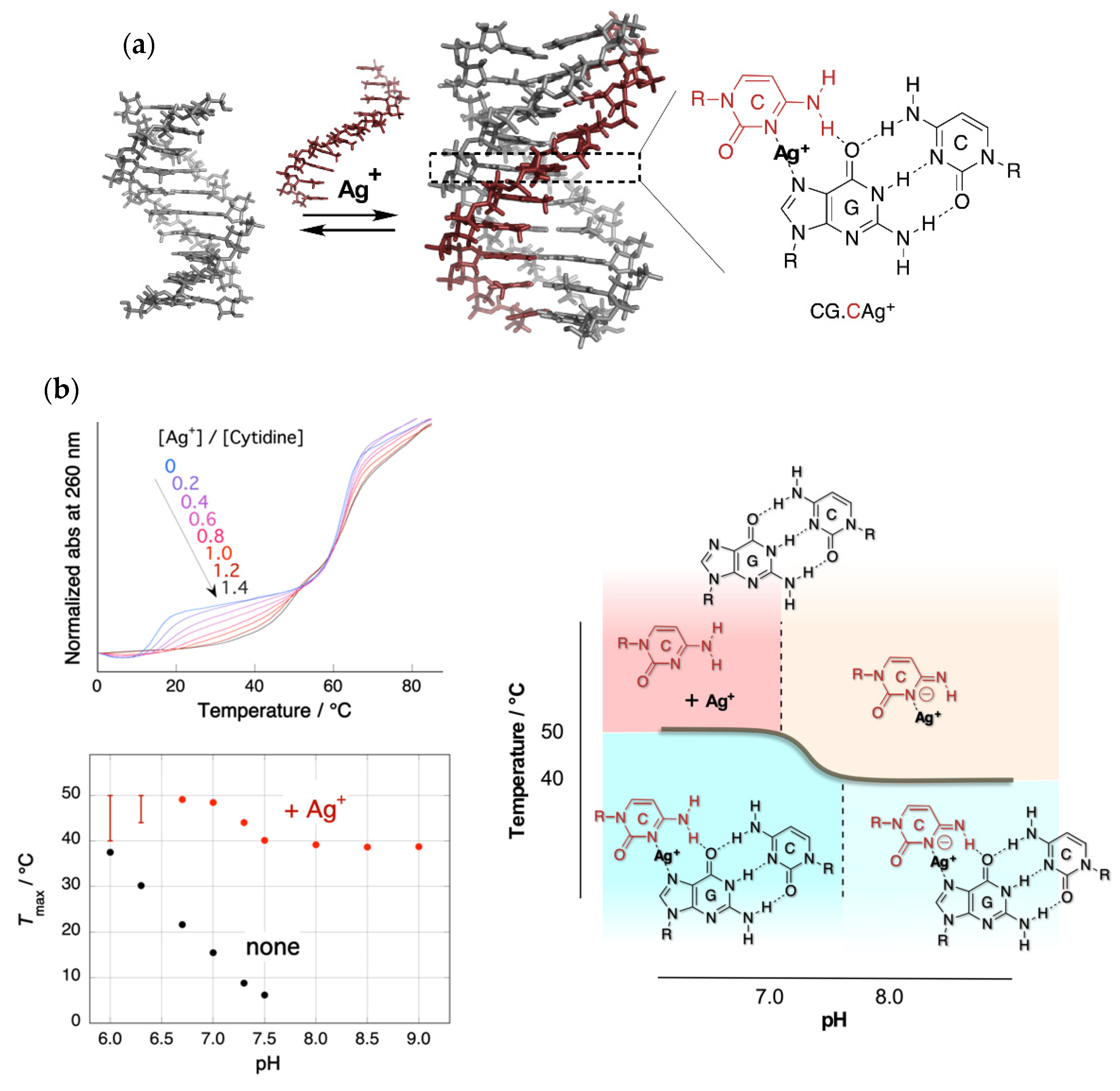
2. Stabilization of a Parallel-Motif DNA Triplex by Silver Ion
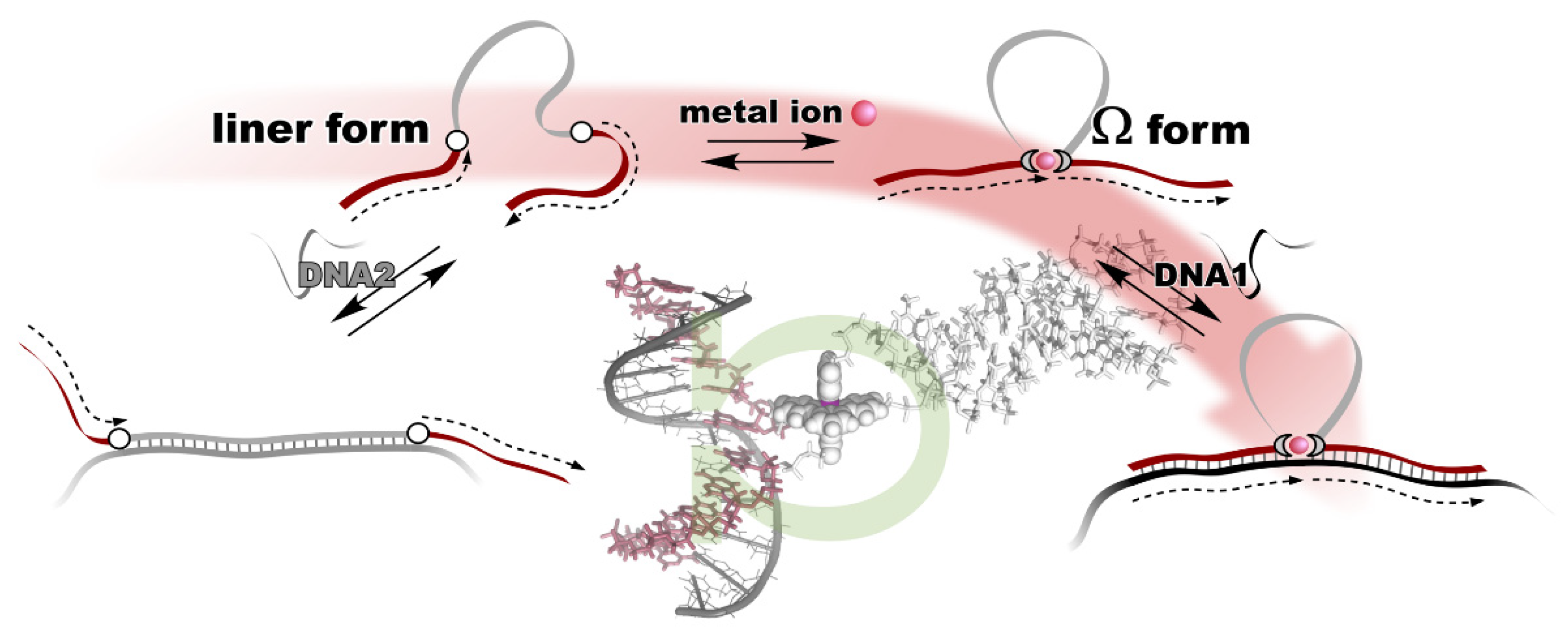
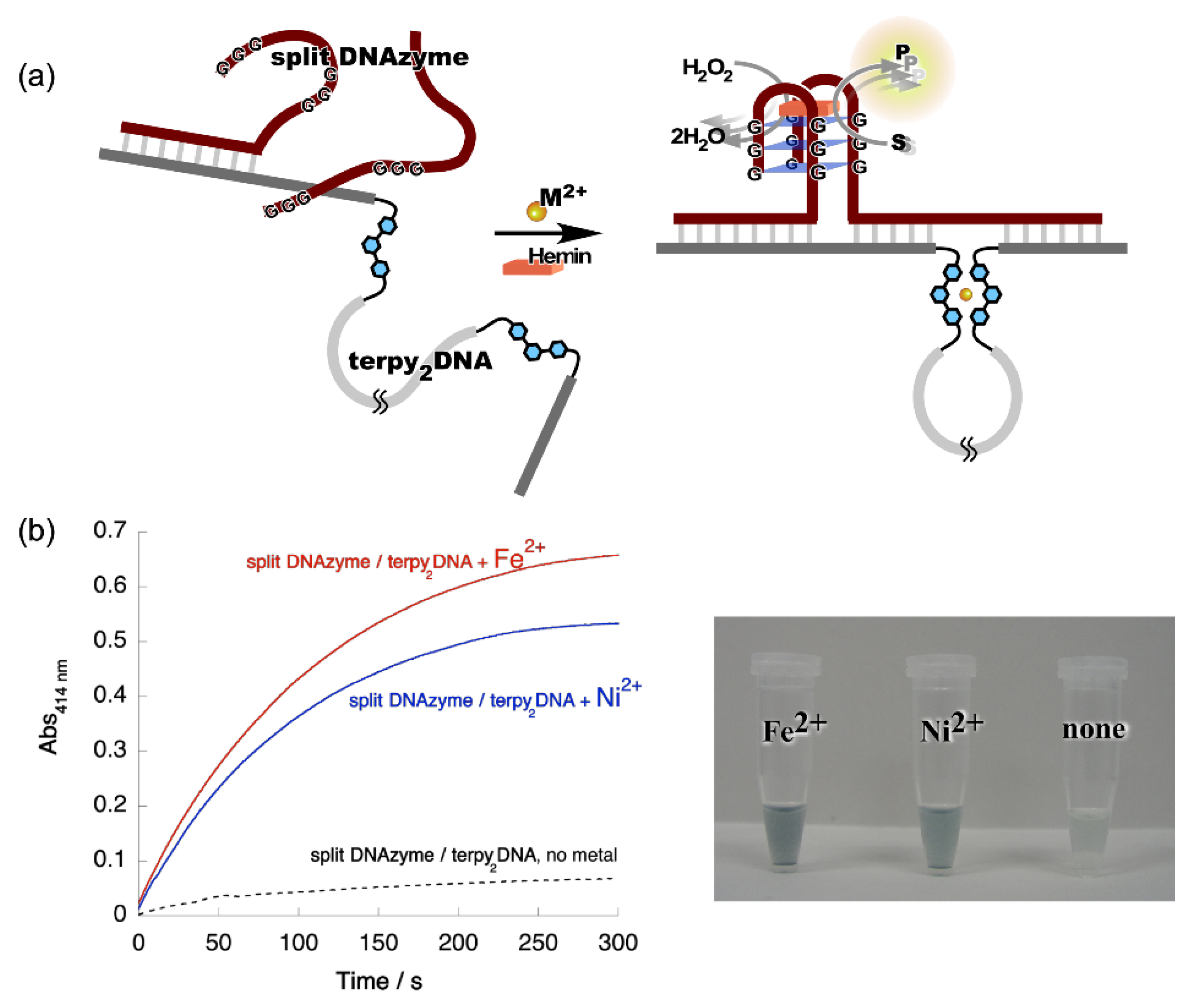
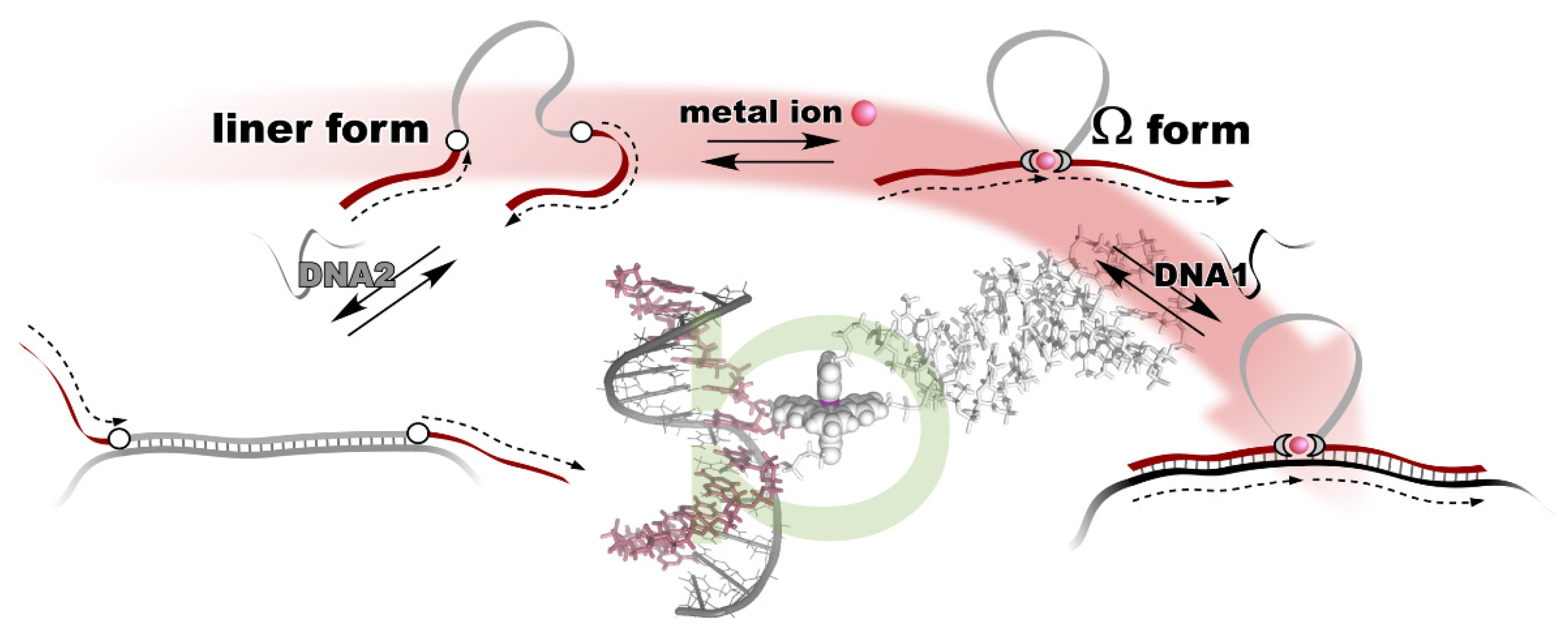
3. Metal Ion-Directed Dynamic Splicing of DNA through Global Conformational Change by Intramolecular Complexation
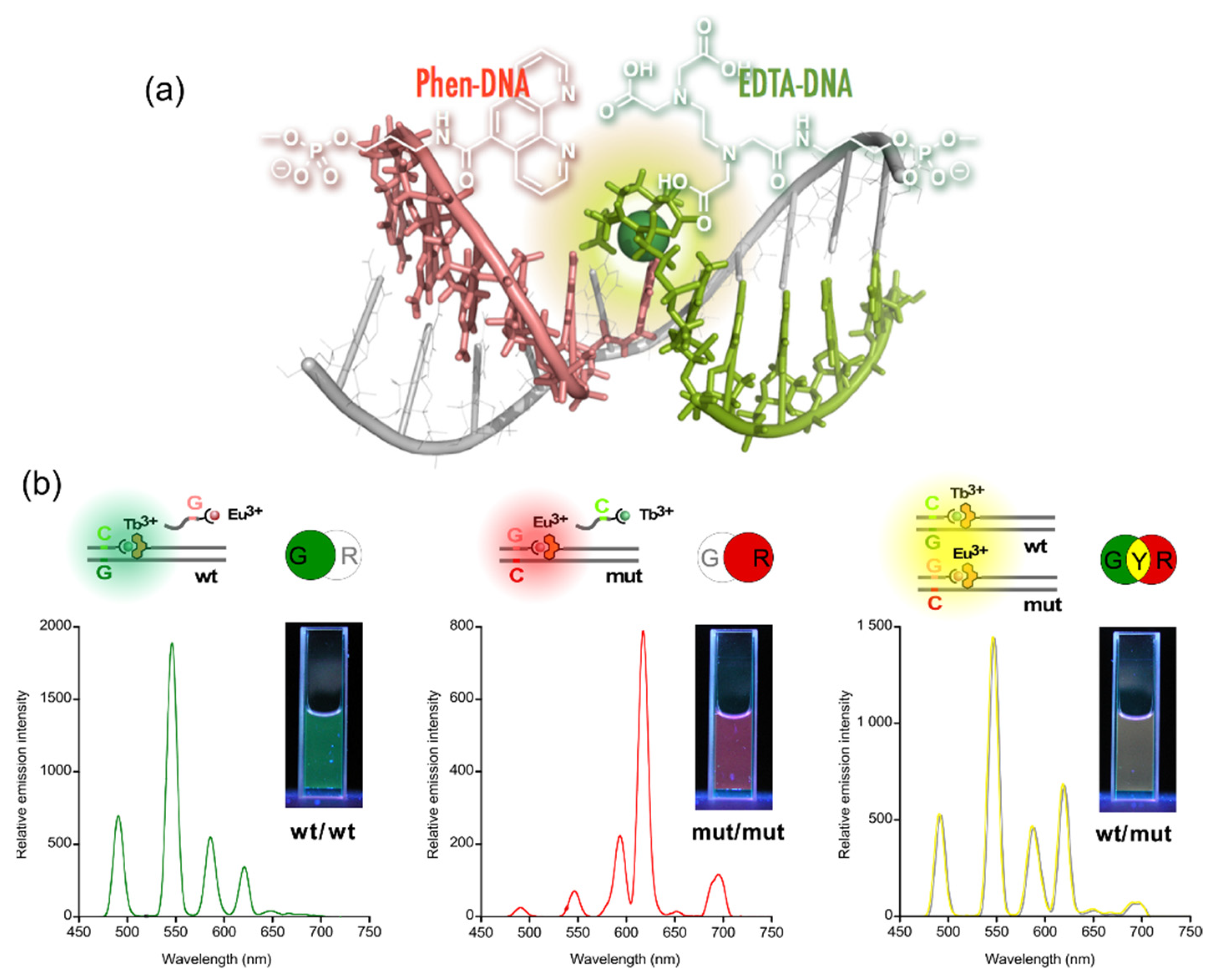
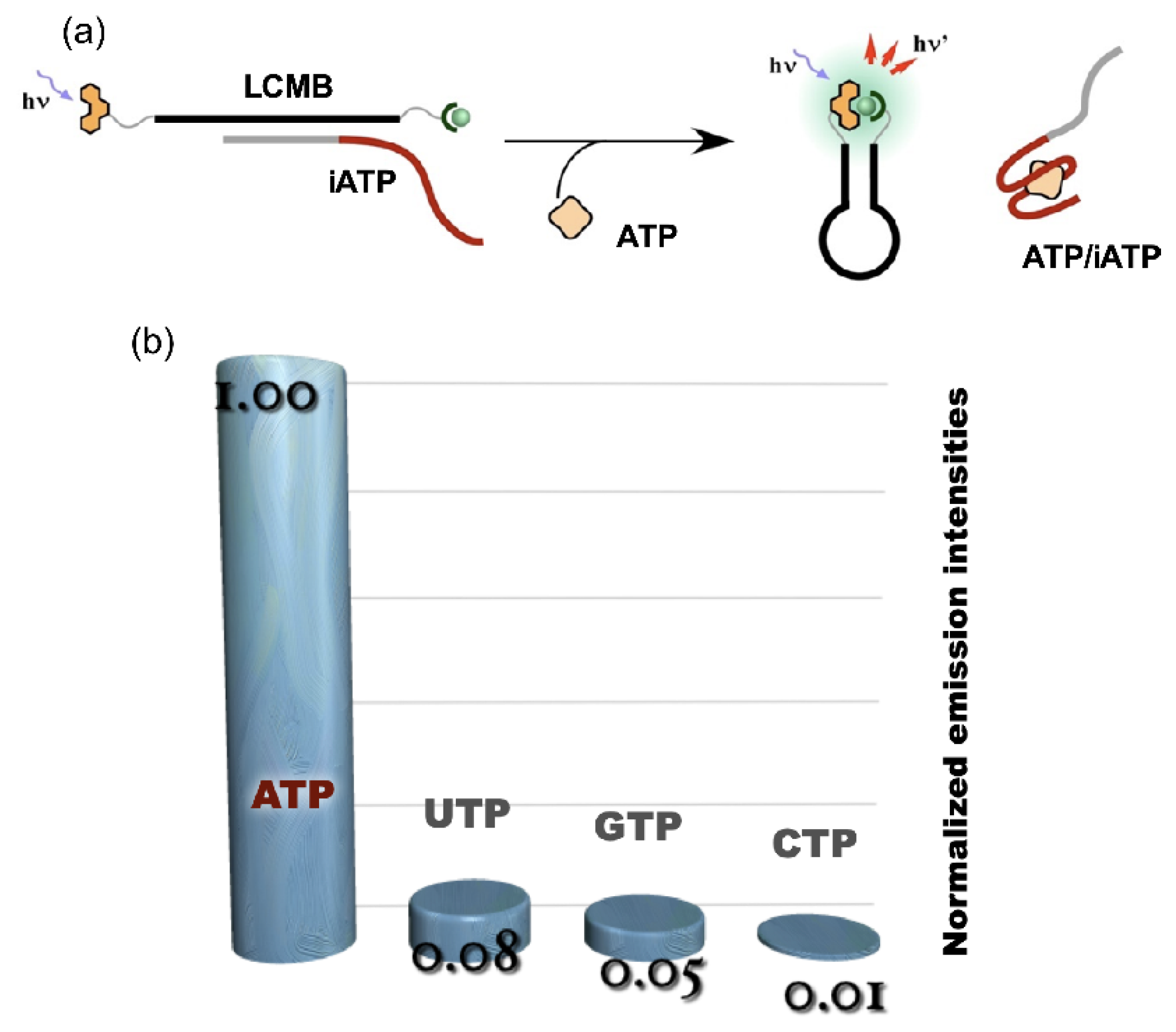
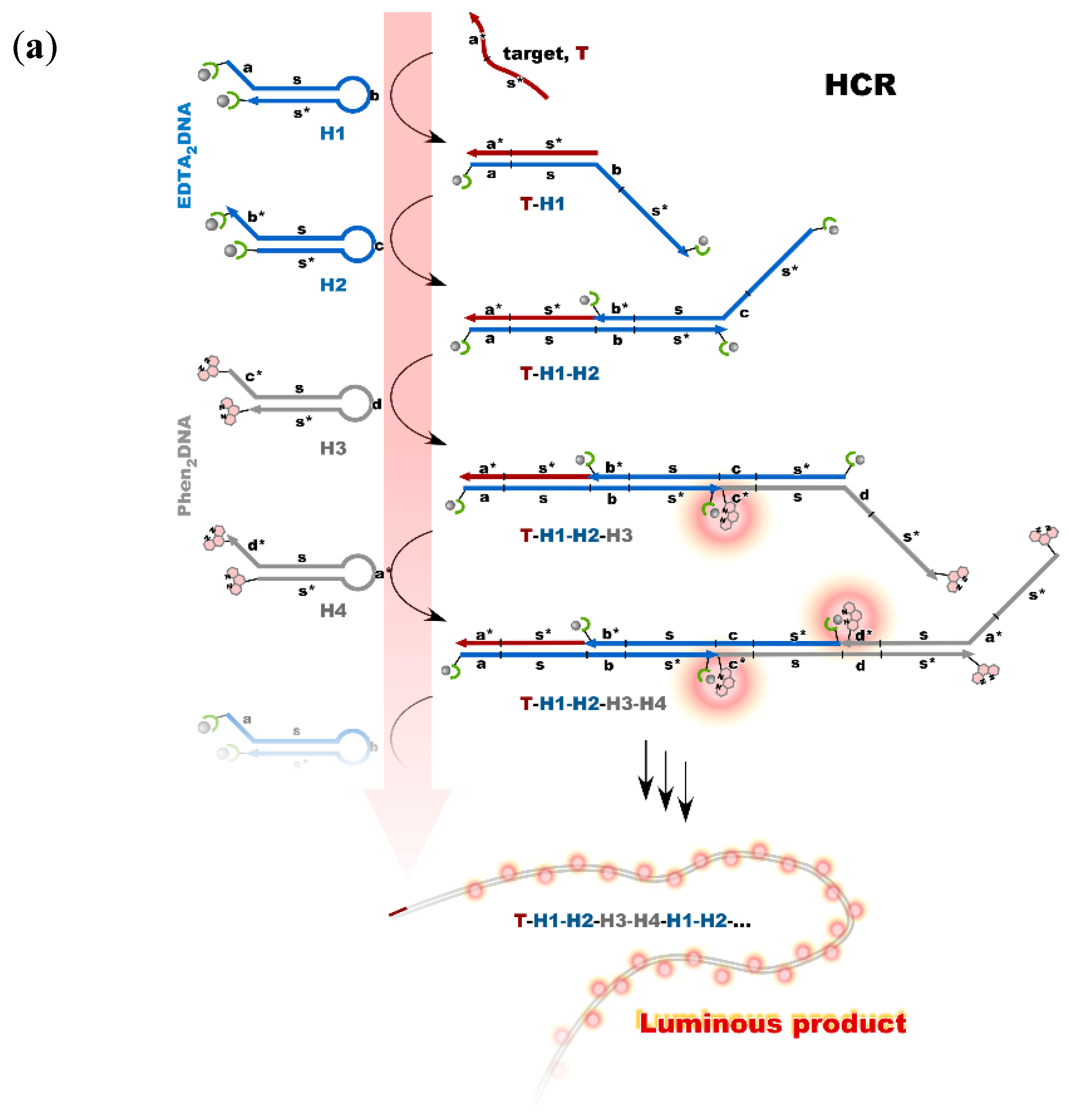
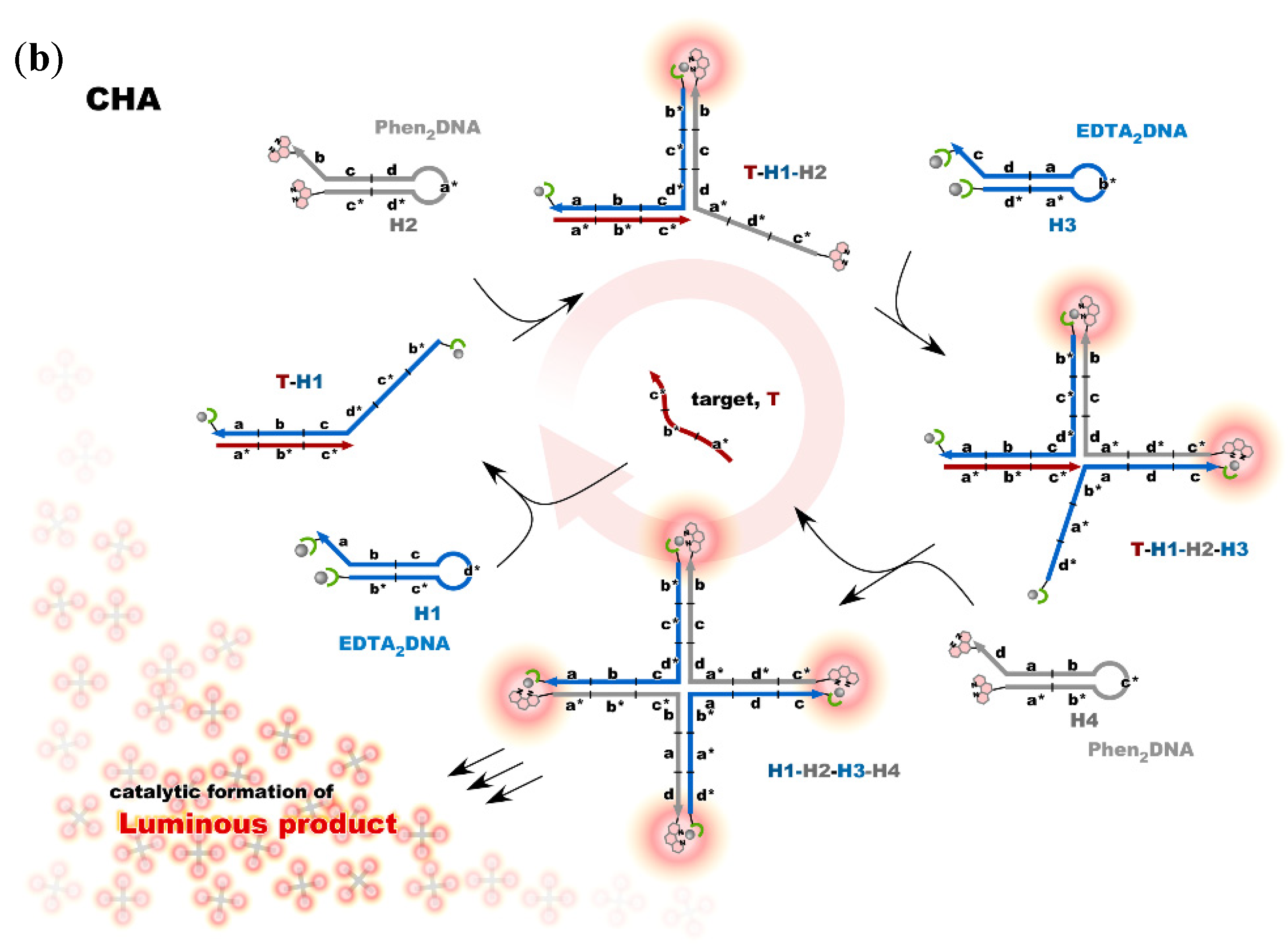
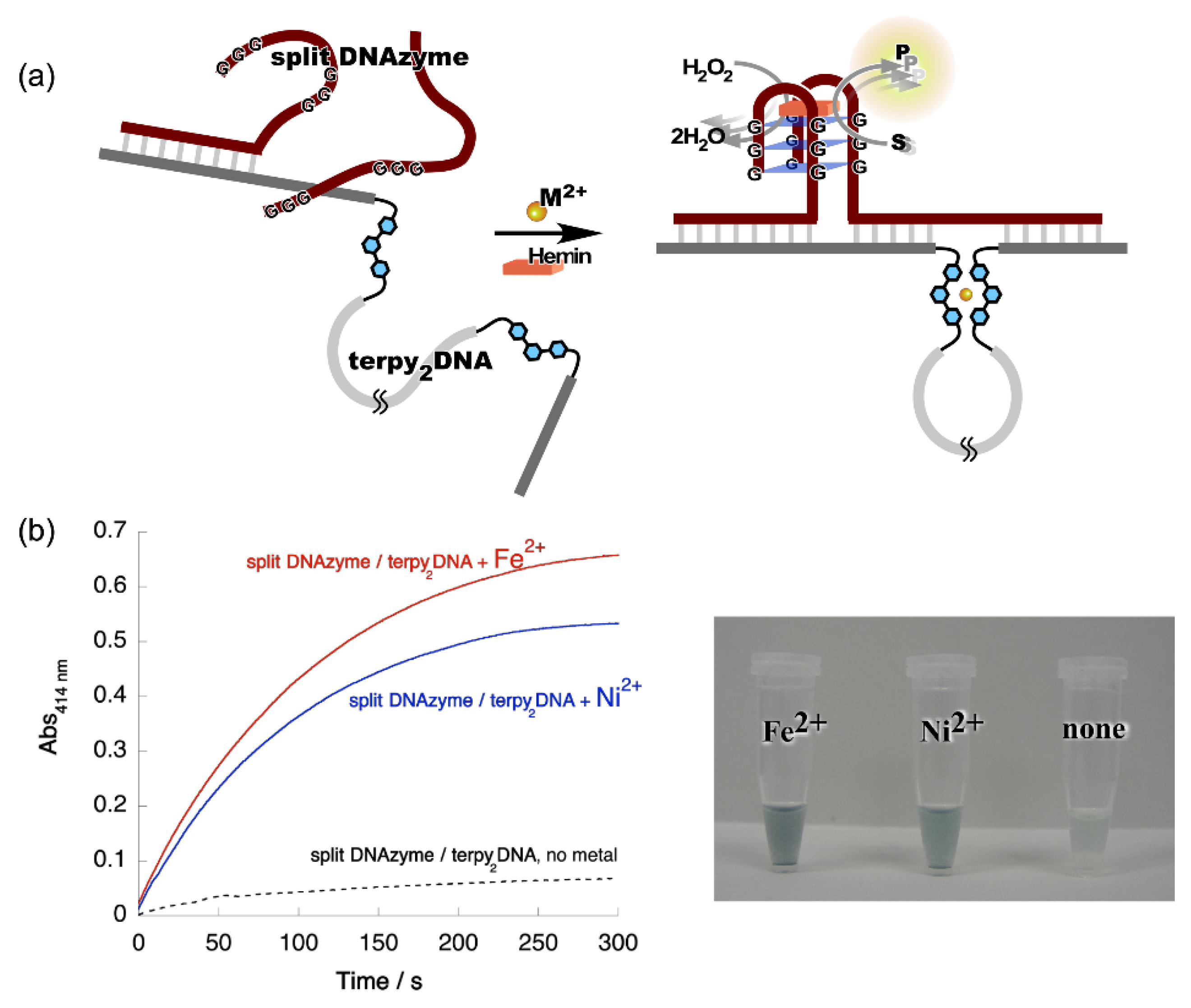
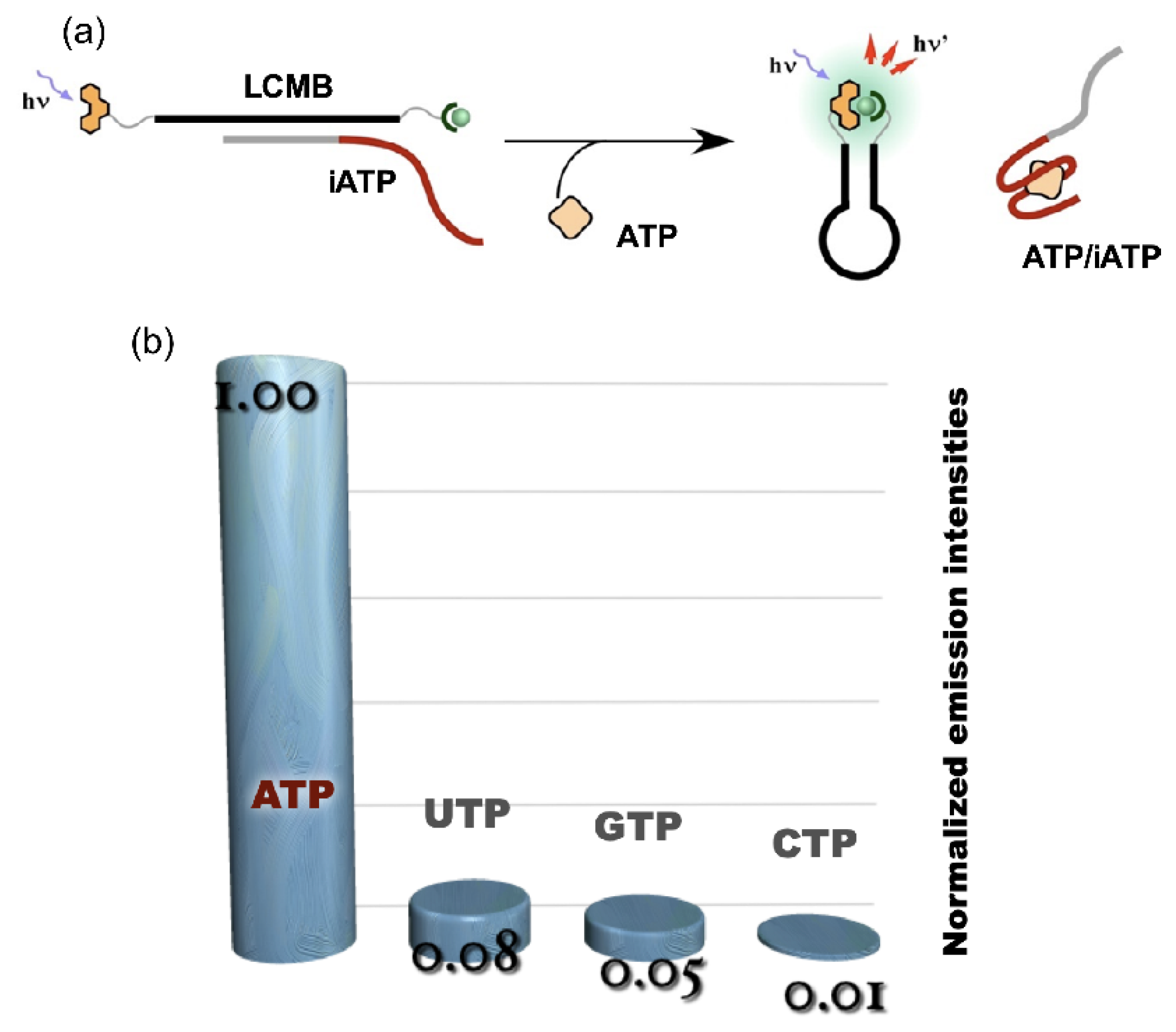
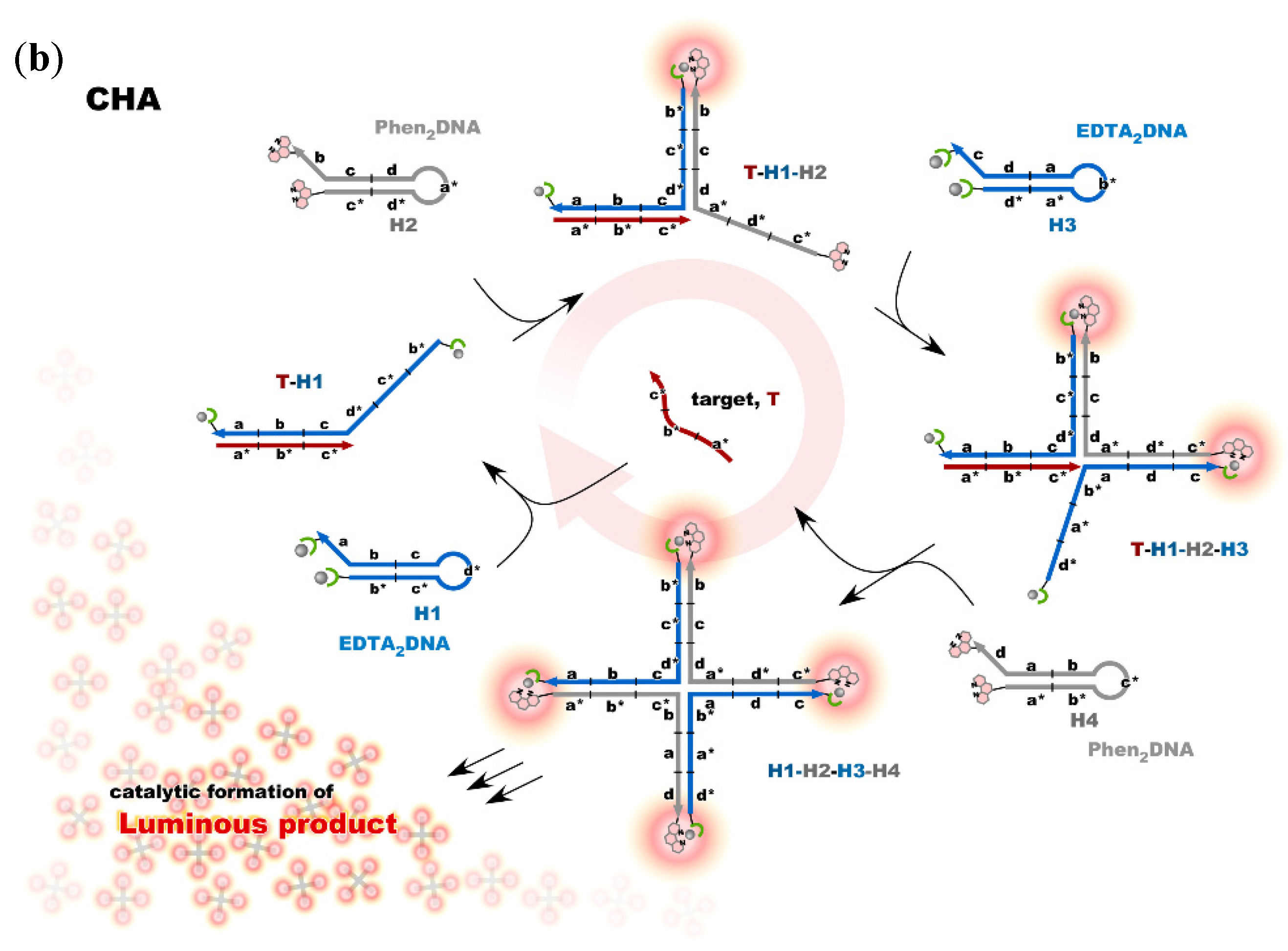
4. Reconstruction of Luminescent Lanthanide Complexes on DNA and Their Analytical Applications
5. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [Google Scholar] [CrossRef] [Green Version]
- Santoro, S.W.; Joyce, G.F.; Sakthivel, K.; Gramatikova, S.; Barbas, C.F. RNA cleavage by a DNA enzyme with expanded chemical functionality. J. Am. Chem. Soc. 2000, 122, 2433–2439. [Google Scholar] [CrossRef]
- Breaker, R.R. Making catalytic DNAs. Science 2000, 290, 2095–2096. [Google Scholar] [CrossRef]
- Seelig, G.; Soloveichik, D.; Zhang, D.Y.; Winfree, E. Enzyme-free nucleic acid logic circuits. Science 2006, 314, 1585–1588. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.U.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef]
- Breslauer, K.J.; Frank, R.; Blöcker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef] [Green Version]
- SantaLucia, J.; Allawi, H.T.; Seneviratne, P.A. Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry 1996, 35, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; SantaLucia, J., Jr.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick base pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, N.; Nakano, S.; Katoh, M.; Matsumura, A.; Nakamuta, H.; Ohmichi, T.; Yoneyama, M.; Sasaki, M. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 1995, 34, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Ihara, T.; Fujii, T.; Mukae, M.; Kitamura, Y.; Jyo, A. Photochemical ligation of DNA conjugates through anthracene cyclodimer formation and its fidelity to the template sequences. J. Am. Chem. Soc. 2004, 126, 8880–8881. [Google Scholar] [CrossRef]
- Ihara, T.; Uemura, A.; Futamura, A.; Shimizu, M.; Baba, N.; Nishizawa, S.; Teramae, N.; Jyo, A. Cooperative DNA probing using a β-cyclodextrin–DNA conjugate and a nucleobase-specific fluorescent ligand. J. Am. Chem. Soc. 2009, 131, 1386–1387. [Google Scholar] [CrossRef] [PubMed]
- Ihara, T.; Wasano, T.; Nakatake, R.; Arslan, P.; Futamura, A.; Jyo, A. Electrochemical signal modulation in homogeneous solutions using the formation of an inclusion complex between ferrocene and β-cyclodextrin on a DNA scaffold. Chem. Commun. 2011, 47, 12388–12390. [Google Scholar] [CrossRef] [PubMed]
- Ihara, T.; Takeda, Y.; Jyo, A. Metal ion-directed cooperative triple helix formation of glutamic acid–oligonucleotide conjugate. J. Am. Chem. Soc. 2001, 123, 1772–1773. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef]
- Silver, G.C.; Sun, J.-S.; Nguyen, C.H.; Boutorine, A.S.; Bisagni, E.; Hélène, C. Stable triple-helical DNA Complexes formed by benzopyridoindole- and benzopyridoquinoxaline-oligonucleotide conjugates. J. Am. Chem. Soc. 1997, 119, 263–268. [Google Scholar] [CrossRef]
- Ihara, T.; Ishi, T.; Araki, N.; Wilson, A.W.; Jyo, A. Silver ion unusually stabilizes the structure of parallel-motif DNA triplex. J. Am. Chem. Soc. 2009, 131, 3826–3827. [Google Scholar] [CrossRef]
- Ono, A.; Torigoe, H.; Tanaka, Y.; Okamoto, I. Binding of metal ion by pyrimidine base pairs in DNA duplexes. Chem. Rev. 2011, 40, 5855–5866. [Google Scholar] [CrossRef] [PubMed]
- Urata, S.; Miyahata, T.; Matsuura, H.; Kitamura, Y.; Ihara, T. Alteration of DNAzyme activity by silver ion. Chem. Lett. 2014, 43, 1020–1022. [Google Scholar] [CrossRef]
- Zhu, D.; Zhu, J.; Zhu, Y.; Wang, L.; Jiang, W. Sensitive detection of transcription factors using an Ag+-stabilized self-assembly triplex DNA molecular switch. Chem. Commun. 2014, 50, 14987–14990. [Google Scholar] [CrossRef]
- Guo, S.; Du, Y.; Yang, X.; Dong, S.; Wang, E. Solid-state label-free integrated aptasensor based on graphene-mesoporous silica–Gold nanoparticle hybrids and silver microspheres. Anal. Chem. 2011, 83, 8035–8040. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Huang, Z.; Ren, J.; Qu, X. Toward site-specific, homogeneous and highly stable fluorescent silver nanoclusters fabrication on triplex DNA scaffolds. Nucleic Acids Res. 2012, 40, e122. [Google Scholar] [CrossRef] [Green Version]
- Ihara, T.; Ohura, H.; Shirahama, C.; Furuzono, T.; Shimada, H.; Matsuura, H.; Kitamura, Y. Metal ion-directed dynamic splicing of DNA through global conformational change by intramolecular complexation. Nat. Commun. 2015, 6, 6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, Y.; Ihara, T.; Tsujimura, Y.; Tazaki, M.; Jyo, A. DNA-templated cooperative formation of the luminous lanthanide complex and its analytical application to gene detection. Chem. Lett. 2005, 34, 1606–1607. [Google Scholar] [CrossRef]
- Ihara, T.; Kitamura, Y.; Tsujimura, Y.; Jyo, A. DNA analysis based on the local structural disruption to the duplexes carrying a luminous lanthanide complex. Anal. Sci. 2011, 27, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.; Ihara, T.; Tsujimura, Y.; Osawa, Y.; Sasahara, D.; Yamamoto, M.; Okada, K.; Tazaki, M.; Jyo, A. Template-directed formation of luminescent lanthanide complexes: Versatile tools for colorimetric identification of single nucleotide polymorphism. J. Inorg. Biochem. 2008, 102, 1921–1931. [Google Scholar] [CrossRef]
- Kitamura, Y.; Yamamoto, S.; Osawa, Y.; Matsuura, H.; Ihara, T. Versatile allosteric molecular devices based on reversible formation of luminous lanthanide complexes. Chem. Commun. 2013, 49, 285–287. [Google Scholar] [CrossRef]
- Kitamura, Y.; Azuma, Y.; Katsuda, Y.; Ihara, T. Catalytic formation of luminescent lanthanide complexes using an entropy-driven DNA circuit. Chem. Commun. 2020, 56, 3863–3866. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Nozaki, A.; Ozaki, R.; Katsuda, Y.; Ihara, T. Catalytic formation of luminescent complex clusters based on autonomous strand exchange reaction of DNA. ACS Appl. Bio Mater. 2019, 2, 2988–2993. [Google Scholar] [CrossRef] [PubMed]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T. A hitchhiker’s guide to click-chemistry with nucleic acids. Chem. Rev. 2021, 121, 7122–7154. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ihara, T.; Kitamura, Y.; Katsuda, Y. Metal Ion-Directed Specific DNA Structures and Their Functions. Life 2022, 12, 686. https://doi.org/10.3390/life12050686
Ihara T, Kitamura Y, Katsuda Y. Metal Ion-Directed Specific DNA Structures and Their Functions. Life. 2022; 12(5):686. https://doi.org/10.3390/life12050686
Chicago/Turabian StyleIhara, Toshihiro, Yusuke Kitamura, and Yousuke Katsuda. 2022. "Metal Ion-Directed Specific DNA Structures and Their Functions" Life 12, no. 5: 686. https://doi.org/10.3390/life12050686
APA StyleIhara, T., Kitamura, Y., & Katsuda, Y. (2022). Metal Ion-Directed Specific DNA Structures and Their Functions. Life, 12(5), 686. https://doi.org/10.3390/life12050686