
Factors That Contribute to hIAPP Amyloidosis in Type 2 Diabetes Mellitus



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Main Text
2.1. The Human Islet Amyloid Polypeptide and Its Link to Diabetes
2.2. The Structure of hIAPP
2.3. Important Residues Involved in hIAPP Aggregation
2.4. The Role of hIAPP Oligomeric Intermediates in T2DM
2.5. Mechanisms of hIAPP Cytotoxicity
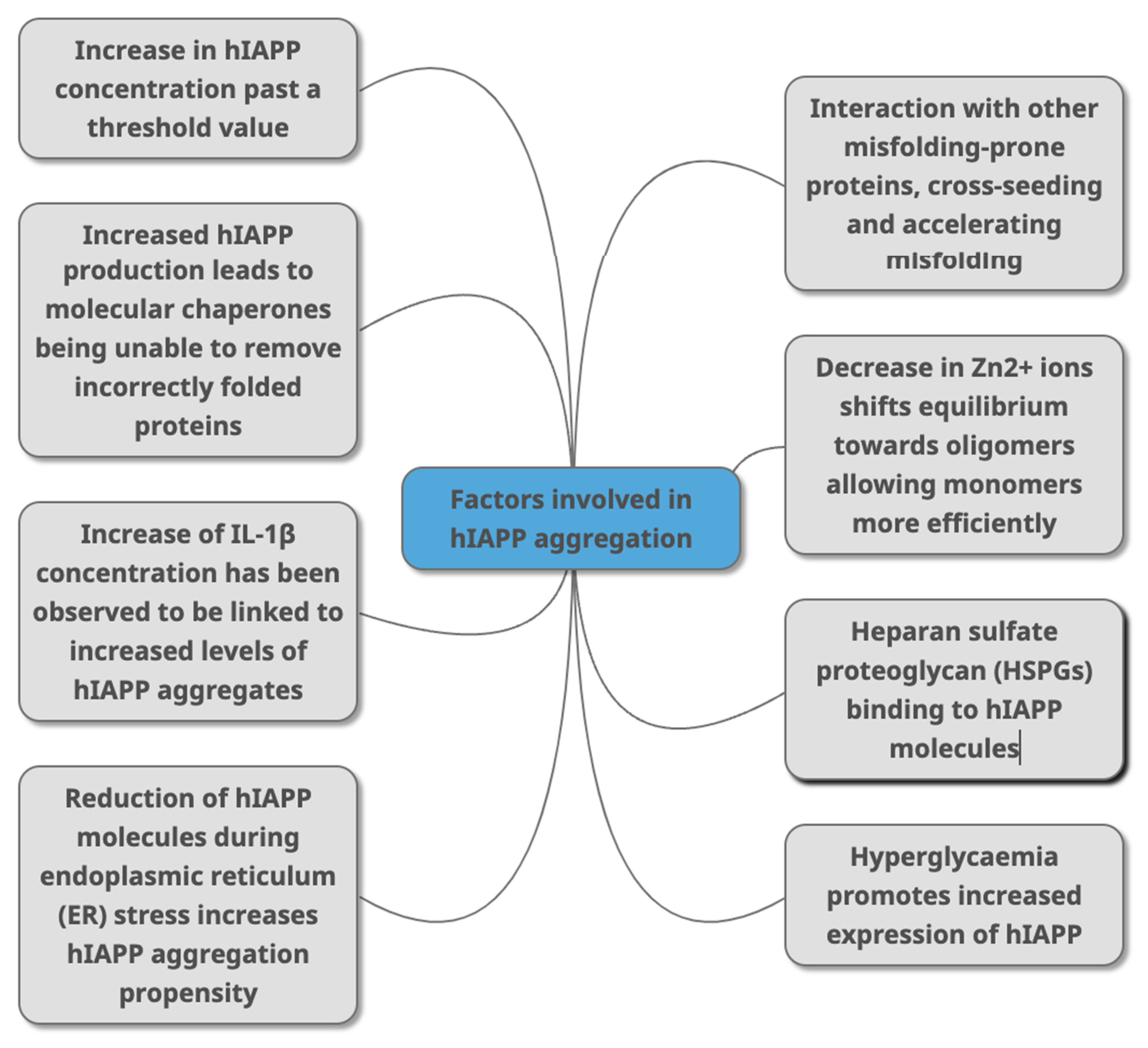
2.6. Acceleration of hIAPP Misfolding and Aggregation
2.6.1. Concentration of hIAPP
2.6.2. Cell Stress
2.6.3. The Role of Chaperones
2.6.4. Heparan Sulphate Proteoglycans
2.6.5. The Immune Response in Amyloidosis
2.6.6. Cross-Seeding of hIAPP and Other Amyloidogenic Proteins
2.6.7. Zinc Ion Concentration
2.7. Type 2 Diabetes Therapeutics
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mittal, K.; Mani, R.; Katare, D. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589. [Google Scholar] [CrossRef] [PubMed]
- Steck, A.; Winter, W. Review on monogenic diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Mei Wong, J.; Sim, Y.; Wong, S.; Mohamed Elhassan, S.; Tan, S.; Ling Lim, G.; Rong Tay, N.; Annan, N.; Bhattamisra, S.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Mack, L.; Tomich, P. Gestational Diabetes. Obstet. Gynecol. Clin. N. Am. 2017, 44, 207–217. [Google Scholar] [CrossRef]
- Nguyen, T.; Ta, Q.; Nguyen, T.; Nguyen, T.; Van Giau, V. Type 3 diabetes and its role implications in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Ceriello, A.; Esposito, K. Glucose metabolism and hyperglycemia. Am. J. Clin. Nutr. 2008, 87, 217S–222S. [Google Scholar] [CrossRef]
- Stratton, I. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int (accessed on 6 April 2022).
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Toplak, H.; Leitner, D.; Harreiter, J.; Hoppichler, F.; Wascher, T.; Schindler, K.; Ludvik, B. “Diabesity”—Adipositas und Typ-2-Diabetes (Update 2019). Wien. Klin. Wochenschr. 2019, 131, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Pulgaron, E.R.; Delamater, A.M. Obesity and Type 2 Diabetes in Children: Epidemiology and Treatment. Curr. Diabetes Rep. 2014, 14, 508. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Nielsen, M.; Fox, M.H. Understanding the Social Factors That Contribute to Diabetes: A Means to Informing Health Care and Social Policies for the Chronically Ill. Perm. J. 2013, 17, 67–72. [Google Scholar] [CrossRef]
- Li, W.-Z.; Stirling, K.; Yang, J.-J.; Zhang, L. Gut microbiota and diabetes: From correlation to causality and mechanism. World J. Diabetes 2020, 11, 293–308. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- Sola, D.; Rossi, L.; Schianca, G.P.C.; Maffioli, P.; Bigliocca, M.; Mella, R.; Corlianò, F.; Fra, G.P.; Bartoli, E.; Derosa, G. State of the art paper Sulfonylureas and their use in clinical practice. Arch. Med. Sci. 2015, 4, 840–848. [Google Scholar] [CrossRef]
- Bishoyi, A.K.; Roham, P.H.; Rachineni, K.; Save, S.; Hazari, M.A.; Sharma, S.; Kumar, A. Human islet amyloid polypeptide (hIAPP)—A curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol. Chem. 2021, 402, 133–153. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Chawla, S.; Guchhait, P. Type-2 diabetes: Current understanding and future perspectives. IUBMB Life 2015, 67, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Hanabusa, T.; Kubo, K.; Oki, C.; Nakano, Y.; Okai, K.; Sanke, T.; Nanjo, K. Islet amyloid polypeptide (IAPP) secretion from islet cells and its plasma concentration in patients with non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1992, 15, 89–96. [Google Scholar] [CrossRef]
- Scollo, F.; La Rosa, C. Amyloidogenic Intrinsically Disordered Proteins: New Insights into Their Self-Assembly and Their Interaction with Membranes. Life 2020, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.J.; Scheen, A.J.; Lefèbvre, P.J. Amylin/islet amyloid polypeptide: Biochemistry, physiology, patho-physiology. Diabete Metab. 1995, 21, 3–25. [Google Scholar] [PubMed]
- Westermark, P.; Engström, U.; Westermark, G.T.; Johnson, K.H.; Permerth, J.; Betsholtz, C. Islet amyloid polypeptide (IAPP) and pro-IAPP immunoreactivity in human islets of Langerhans. Diabetes Res. Clin. Pract. 1989, 7, 219–226. [Google Scholar] [CrossRef]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef]
- Wang, J.; Xu, J.; Finnerty, J.; Furuta, M.; Steiner, D.F.; Verchere, C.B. The Prohormone Convertase Enzyme 2 (PC2) Is Essential for Processing Pro-Islet Amyloid Polypeptide at the NH2-Terminal Cleavage Site. Diabetes 2001, 50, 534–539. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef]
- Cao, P.; Abedini, A.; Raleigh, D.P. Aggregation of islet amyloid polypeptide: From physical chemistry to cell biology. Curr. Opin. Struct. Biol. 2012, 23, 82–89. [Google Scholar] [CrossRef]
- Mulder, H.; Ahren, B.; Sundler, F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. Metab. 1996, 271, E1008–E1014. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H.; Gebre-Medhin, S.; Betsholtz, C.; Sundler, F.; Ahrén, B. Islet amyloid polypeptide (amylin)-deficient mice develop a more severe form of alloxan-induced diabetes. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E684–E691. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Sonar, K.; Bharadwaj, P.; Deplazes, E.; Mancera, R.L. Characterisation of the Structure and Oligomerisation of Islet Amyloid Polypeptides (IAPP): A Review of Molecular Dynamics Simulation Studies. Molecules 2018, 23, 2142. [Google Scholar] [CrossRef] [PubMed]
- Higham, C.E.; Jaikaran, E.T.; Fraser, P.E.; Gross, M.; Clark, A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000, 470, 55–60. [Google Scholar] [CrossRef]
- Bedrood, S.; Li, Y.; Isas, J.M.; Hegde, B.G.; Baxa, U.; Haworth, I.S.; Langen, R. Fibril Structure of Human Islet Amyloid Polypeptide. J. Biol. Chem. 2012, 287, 5235–5241. [Google Scholar] [CrossRef]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations That Potentially Seed Aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef]
- Fu, Z.; Aucoin, D.; Davis, J.; Van Nostrand, W.E.; Smith, S.O. Mechanism of Nucleated Conformational Conversion of Aβ42. Biochemistry 2015, 54, 4197–4207. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLOS Comput. Biol. 2016, 12, e1005067. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.; Sawaya, M.; Ge, P.; Eisenberg, D. Cryo-EM structure and inhibitor design of humanIAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 2020, 27, 653–659. [Google Scholar] [CrossRef]
- Gallardo, R.; Iadanza, M.G.; Xu, Y.; Heath, G.R.; Foster, R.; Radford, S.E.; Ranson, N.A. Fibril structures of diabetes-related amylin variants reveal a basis for surface-templated assembly. Nat. Struct. Mol. Biol. 2020, 27, 1048–1056. [Google Scholar] [CrossRef]
- Röder, C.; Kupreichyk, T.; Gremer, L.; Schäfer, L.U.; Pothula, K.R.; Ravelli, R.B.G.; Willbold, D.; Hoyer, W.; Schröder, G.F. Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat. Struct. Mol. Biol. 2020, 27, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, D.; Zhu, X.; Wang, Y.; Zhu, P. Structural characterization and cryo-electron tomography analysis of human islet amyloid polypeptide suggest a synchronous process of the hIAPP1−37 amyloid fibrillation. Biochem. Biophys. Res. Commun. 2020, 533, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Abskharon, R.; Saelices, L.; Nguyen, B.A.; Lu, J.; Murray, K.A.; Kandeel, F.; Eisenberg, D.S. Cryo-EM structures of hIAPP fibrils seeded by patient-extracted fibrils reveal new polymorphs and conserved fibril cores. Nat. Struct. Mol. Biol. 2021, 28, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Li, M.S.; Klimov, D.K.; Straub, J.E.; Thirumalai, D. Probing the mechanisms of fibril formation using lattice models. J. Chem. Phys. 2008, 129, 175101. [Google Scholar] [CrossRef]
- Brender, J.; Dürr, U.H.; Heyl, D.; Budarapu, M.B.; Ramamoorthy, A. Membrane fragmentation by an amyloidogenic fragment of human Islet Amyloid Polypeptide detected by solid-state NMR spectroscopy of membrane nanotubes. Biochim. Biophys. Acta (BBA) 2007, 1768, 2026–2029. [Google Scholar] [CrossRef][Green Version]
- Apostolidou, M.; Jayasinghe, S.A.; Langen, R. Structure of α-Helical Membrane-bound Human Islet Amyloid Polypeptide and Its Implications for Membrane-mediated Misfolding. J. Biol. Chem. 2008, 283, 17205–17210. [Google Scholar] [CrossRef]
- Zhang, X.; Clair, J.R.S.; London, E.; Raleigh, D.P. Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry 2017, 56, 376–390. [Google Scholar] [CrossRef]
- Sasahara, K. Membrane-mediated amyloid deposition of human islet amyloid polypeptide. Biophys. Rev. 2018, 10, 453–462. [Google Scholar] [CrossRef][Green Version]
- Caillon, L.; Hoffmann, A.R.F.; Botz, A.; Khemtemourian, L. Molecular Structure, Membrane Interactions, and Toxicity of the Islet Amyloid Polypeptide in Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 5639875. [Google Scholar] [CrossRef]
- Bhowmick, D.C.; Kudaibergenova, Z.; Burnett, L.; Jeremic, A.M. Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas. Molecules 2022, 27, 1021. [Google Scholar] [CrossRef]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.-J.; Trikha, S.; Jeremic, A.M. Cholesterol Regulates Assembly of Human Islet Amyloid Polypeptide on Model Membranes. J. Mol. Biol. 2009, 393, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, O.; Höhn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of Amylin by Insulin-degrading Enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Miranda, H.V. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An Insulin-Degrading Enzyme Inhibitor Decreases Amylin Degradation, Increases Amylin-Induced Cytotoxicity, and Increases Amyloid Formation in Insulinoma Cell Cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef]
- Hogan, M.F.; Zeman-Meier, D.; Zraika, S.; Templin, A.; Mellati, M.; Hull, R.L.; Leissring, M.A.; Kahn, S.E. Inhibition of Insulin-Degrading Enzyme Does Not Increase Islet Amyloid Deposition in Vitro. Endocrinology 2016, 157, 3462–3468. [Google Scholar] [CrossRef]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.-J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef]
- Tang, W.-J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef]
- Luca, S.; Yau, W.-M.; Leapman, R.; Tycko, R. Peptide Conformation and Supramolecular Organization in Amylin Fibrils: Constraints from Solid-State NMR. Biochemistry 2007, 46, 13505–13522. [Google Scholar] [CrossRef]
- Wiltzius, J.J.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin). Protein Sci. 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-cell assassin: IAPP cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.Q.; McGovern, M.; Chiu, C.-C.; De Pablo, J.J. Secondary Structure of Rat and Human Amylin across Force Fields. PLoS ONE 2015, 10, e0134091. [Google Scholar] [CrossRef] [PubMed]
- Wineman-Fisher, V.; Atsmon-Raz, Y.; Miller, Y. Orientations of Residues along the β-Arch of Self-Assembled Amylin Fibril-Like Structures Lead to Polymorphism. Biomacromolecules 2015, 16, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Butler, P.C. β-Cell Deficit Due to Increased Apoptosis in the Human Islet Amyloid Polypeptide Transgenic (HIP) Rat Recapitulates the Metabolic Defects Present in Type 2 Diabetes. Diabetes 2006, 55, 2106–2114. [Google Scholar] [CrossRef]
- Krotee, P.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Hattne, J.; Nannenga, B.; Oskarsson, M.E.; Philipp, S.; et al. Atomic structures of fibrillar segments of hIAPP suggest tightly mated β-sheets are important for cytotoxicity. eLife 2017, 6, e19273. [Google Scholar] [CrossRef]
- Zeman-Meier, D.; Entrup, L.; Templin, A.; Hogan, M.F.; Mellati, M.; Zraika, S.; Hull, R.L.; Kahn, S.E. The S20G substitution in hIAPP is more amyloidogenic and cytotoxic than wild-type hIAPP in mouse islets. Diabetologia 2016, 59, 2166–2171. [Google Scholar] [CrossRef]
- Ridgway, Z.; Zhang, X.; Wong, A.G.; Abedini, A.; Schmidt, A.M.; Raleigh, D.P. Analysis of the Role of the Conserved Disulfide in Amyloid Formation by Human Islet Amyloid Polypeptide in Homogeneous and Heterogeneous Environments. Biochemistry 2018, 57, 3065–3074. [Google Scholar] [CrossRef]
- Altamirano-Bustamante, M.M.; Altamirano-Bustamante, N.F.; Larralde-Laborde, M.; Lara-Martínez, R.; Leyva-García, E.; Garrido-Magaña, E.; Rojas, G.; Jiménez-García, L.F.; Monsalve, M.C.R.; Altamirano, P.; et al. Unpacking the aggregation-oligomerization-fibrillization process of naturally-occurring hIAPP amyloid oligomers isolated directly from sera of children with obesity or diabetes mellitus. Sci. Rep. 2019, 9, 18465. [Google Scholar] [CrossRef]
- Burillo, J.; Fernández-Rhodes, M.; Piquero, M.; López-Alvarado, P.; Menéndez, J.; Jiménez, B.; González-Blanco, C.; Marqués, P.; Guillén, C.; Benito, M. Human amylin aggregates release within exosomes as a protective mechanism in pancreatic β cells: Pancreatic β-hippocampal cell communication. Biochim. Biophys. Acta 2021, 1868, 118971. [Google Scholar] [CrossRef]
- Hiddinga, H.J.; Eberhardt, N.L. Intracellular Amyloidogenesis by Human Islet Amyloid Polypeptide Induces Apoptosis in COS-1 Cells. Am. J. Pathol. 1999, 154, 1077–1088. [Google Scholar] [CrossRef]
- Bag, N.; Ali, A.; Chauhan, V.S.; Wohland, T.; Mishra, A. Membrane destabilization by monomeric hIAPP observed by imaging fluorescence correlation spectroscopy. Chem. Commun. 2013, 49, 9155–9157. [Google Scholar] [CrossRef]
- Pilkington, E.; Gurzov, E.; Kakinen, A.; A Litwak, S.; Stanley, W.J.W.; Davis, T.P.T.; Ke, P.C. Pancreatic β-Cell Membrane Fluidity and Toxicity Induced by Human Islet Amyloid Polypeptide Species. Sci. Rep. 2016, 6, 21274. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.M.; Khemtémourian, L.; Kleijer, C.C.; Meeldijk, H.J.D.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Höppener, J.W.M. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Kudva, Y.C.; Mueske, C.; Butler, P.C.; Eberhardt, N.L. A novel assay in vitro of human islet amyloid polypeptide amyloidogenesis and effects of insulin secretory vesicle peptides on amyloid formation. Biochem. J. 1998, 331, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.; Krishnamoorthy, J.; Sciacca, M.F.M.; Vivekanandan, S.; D’Urso, L.; Chen, J.; La Rosa, C.; Ramamoorthy, A. Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 2015, 119, 2886–2896. [Google Scholar] [CrossRef] [PubMed]
- A Kassir, A.; Upadhyay, A.K.; Lim, T.J.; Moossa, A.R.; Olefsky, J.M. Lack of Effect of Islet Amyloid Polypeptide in Causing Insulin Resistance in Conscious Dogs During Euglycemic Clamp Studies. Diabetes 1991, 40, 998–1004. [Google Scholar] [CrossRef]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2007, 29, 42–61. [Google Scholar] [CrossRef]
- Camargo, D.C.R.; Tripsianes, K.; Buday, K.; Franko, A.; Göbl, C.; Hartlmüller, C.; Sarkar, R.; Aichler, M.; Mettenleiter, G.; Schulz, M.; et al. The redox environment triggers conformational changes and aggregation of hIAPP in Type II Diabetes. Sci. Rep. 2017, 7, 44041. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.W.; Khanolkar, M.P.; Bain, S.C. The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis 2009, 202, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.T.; Poitout, V. β-Cell Glucose Toxicity, Lipotoxicity, and Chronic Oxidative Stress in Type 2 Diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Morón, E.B.; Abad-Jiménez, Z.; De Marañón, A.M.; Iannantuoni, F.; López, E.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ni, B.; Zhang, F.; Hu, Y.; Zhao, J. High Glucose-Induced Oxidative Stress Mediates Apoptosis and Extracellular Matrix Metabolic Imbalances Possibly via p38 MAPK Activation in Rat Nucleus Pulposus Cells. J. Diabetes Res. 2016, 2016, 3765173. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef]
- Fiorentino, T.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Mele, B.H.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Saibil, H.R. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Erickson, R.R.; Dunning, L.M.; Holtzman, J.L. The Effect of Aging on the Chaperone Concentrations in the Hepatic, Endoplasmic Reticulum of Male Rats: The Possible Role of Protein Misfolding Due to the Loss of Chaperones in the Decline in Physiological Function Seen With Age. J. Gerontol. Ser. A 2006, 61, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Cadavez, L.; Montane, J.; Alcarraz-Vizán, G.; Visa, M.; Vidal-Fàbrega, L.; Servitja, J.-M.; Novials, A. Chaperones Ameliorate Beta Cell Dysfunction Associated with Human Islet Amyloid Polypeptide Overexpression. PLoS ONE 2014, 9, e101797. [Google Scholar] [CrossRef]
- Fernández-Gómez, I.; Sablón-Carrazana, M.; Bencomo-Martínez, A.; Domínguez, G.; Lara-Martínez, R.; Altamirano-Bustamante, N.F.; Jiménez-García, L.F.; Pasten-Hidalgo, K.; Castillo-Rodríguez, R.A.; Altamirano, P.; et al. Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules 2018, 23, 686. [Google Scholar] [CrossRef] [PubMed]
- Nakhjavani, M.; Morteza, A.; Khajeali, L.; Esteghamati, A.; Khalilzadeh, O.; Asgarani, F.; Outeiro, T.F. Increased serum HSP70 levels are associated with the duration of diabetes. Cell Stress Chaperones 2010, 15, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Ladjimi, M.; Chaari, A.; Eliezer, D. Inhibition of Human Islet Amyloid Polypeptide Aggregation in Type 2 Diabetes by Hsp70 Molecular Chaperones. In Proceedings of the Qatar Foundation Annual Research Conference Proceedings 2016, Doha, Qatar, 22–23 March 2016; p. HBPP2702. [Google Scholar] [CrossRef]
- Chilukoti, N.; Sil, T.B.; Sahoo, B.; Deepa, S.; Cherakara, S.; Maddheshiya, M.; Garai, K. Hsp70 Inhibits Aggregation of IAPP by Binding to the Heterogeneous Prenucleation Oligomers. Biophys. J. 2021, 120, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Mulyani, W.R.W.; Sanjiwani, M.I.D.; Sandra, S.; Prabawa, I.P.Y.; Lestari, A.A.W.; Wihandani, D.M.; Suastika, K.; Saraswati, M.R.; Bhargah, A.; Manuaba, I.B.A.P. Chaperone-Based Therapeutic Target Innovation: Heat Shock Protein 70 (HSP70) for Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 559–568. [Google Scholar] [CrossRef]
- Condomitti, G.; De Wit, J. Heparan Sulfate Proteoglycans as Emerging Players in Synaptic Specificity. Front. Mol. Neurosci. 2018, 11, 14. [Google Scholar] [CrossRef]
- Templin, A.T.; Mellati, M.; Soininen, R.; Hogan, M.F.; Esser, N.; Castillo, J.J.; Zraika, S.; E Kahn, S.; Hull, R.L. Loss of perlecan heparan sulfate glycosaminoglycans lowers body weight and decreases islet amyloid deposition in human islet amyloid polypeptide transgenic mice. Protein Eng. Des. Sel. 2019, 32, 95–102. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- O’Neill, C.M.; Lu, C.; Corbin, K.L.; Sharma, P.R.; Dula, S.B.; Carter, J.D.; Ramadan, J.W.; Xin, W.; Lee, J.K.; Nunemaker, C.S. Circulating Levels of IL-1B+IL-6 Cause ER Stress and Dysfunction in Islets From Prediabetic Male Mice. Endocrinology 2013, 154, 3077–3088. [Google Scholar] [CrossRef]
- Templin, A.T.; Mellati, M.; Meier, D.T.; Esser, N.; Hogan, M.F.; Castillo, J.J.; Akter, R.; Raleigh, D.P.; Zraika, S.; Hull, R.L.; et al. Low concentration IL-1β promotes islet amyloid formation by increasing hIAPP release from humanised mouse islets in vitro. Diabetologia 2020, 63, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Dalmas, E.; Böni-Schnetzler, M.; Donath, M.Y. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends Endocrinol. Metab. 2015, 26, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Chylikova, J.; Dvorackova, J.; Tauber, Z.; Kamarad, V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed. Pap. 2018, 162, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Y.; Zhang, D.; Liu, Y.; He, J.; Chang, Y.; Zheng, J. Amyloid cross-seeding between Aβ and hIAPP in relation to the pathogenesis of Alzheimer and type 2 diabetes. Chin. J. Chem. Eng. 2021, 30, 225–235. [Google Scholar] [CrossRef]
- Hu, R.; Zhang, M.; Chen, H.; Jiang, B.; Zheng, J. Cross-Seeding Interaction between β-Amyloid and Human Islet Amyloid Polypeptide. ACS Chem. Neurosci. 2015, 6, 1759–1768. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Ren, B.; Chen, H.; Jiang, B.; Ma, J.; Zheng, J. Molecular Understanding of Aβ-hIAPP Cross-Seeding Assemblies on Lipid Membranes. ACS Chem. Neurosci. 2016, 8, 524–537. [Google Scholar] [CrossRef]
- Atsmon-Raz, Y.; Miller, Y. Co-Aggregation of Alpha-Synuclein with Amylin(HIAPP) Leads to an Increased Risk in Type ii Diabetes Patients for Developing Parkinson’s Disease. Biophys. J. 2015, 108, 524a. [Google Scholar] [CrossRef][Green Version]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Nanga, R.P.R.; Popovych, N.; Bea, R.D.L.S.; Vivekanandan, S.; Marsh, E.N.G.; Ramamoorthy, A. Role of Zinc in Human Islet Amyloid Polypeptide Aggregation. J. Am. Chem. Soc. 2010, 132, 8973–8983. [Google Scholar] [CrossRef]
- Govindan, P.N.; Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef]
- Hemmingsen, B.; Gimenez-Perez, G.; Mauricio, D.; Figuls, M.R.I.; Metzendorf, M.-I.; Richter, B. Diet, physical activity or both for prevention or delay of type 2 diabetes mellitus and its associated complications in people at increased risk of developing type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2017, 2017, CD003054. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Ma, L.; Yang, C.; Zheng, J.; Chen, Y.; Xiao, Y.; Huang, K. Non-polyphenolic natural inhibitors of amyloid aggregation. Eur. J. Med. Chem. 2020, 192, 112197. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-Z.; Ji, J.-G.; Zheng, L.-J.; Shen, J.; Li, X.-Y.; Zhang, Q.; Bai, X.; Wang, Q.-S. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 2018, 13, 2005. [Google Scholar] [CrossRef]
- Lv, W.; Zhang, J.; Jiao, A.; Wang, B.; Chen, B.; Lin, J. Resveratrol attenuates hIAPP amyloid formation and restores the insulin secretion ability in hIAPP-INS1 cell line via enhancing autophagy. Can. J. Physiol. Pharmacol. 2019, 97, 82–89. [Google Scholar] [CrossRef]
- Terry, C. Insights from nature: A review of natural compounds that target protein misfolding in vivo. Curr. Res. Biotechnol. 2020, 2, 131–144. [Google Scholar] [CrossRef]
- Bahramikia, S.; Yazdanparast, R. Inhibition of human islet amyloid polypeptide or amylin aggregation by two manganese-salen derivatives. Eur. J. Pharmacol. 2013, 707, 17–25. [Google Scholar] [CrossRef]
- Gorogawa, S.-I.; Kajimoto, Y.; Umayahara, Y.; Kaneto, H.; Watada, H.; Kuroda, A.; Kawamori, D.; Yasuda, T.; Matsuhisa, M.; Yamasaki, Y.; et al. Probucol preserves pancreatic β-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res. Clin. Pract. 2002, 57, 1–10. [Google Scholar] [CrossRef]
- Lo, M.C.; Lansang, M.C. Recent and Emerging Therapeutic Medications in Type 2 Diabetes Mellitus. Am. J. Ther. 2013, 20, 638–653. [Google Scholar] [CrossRef]
- Bram, Y.; Peled, S.; Brahmachari, S.; Harlev, M.; Gazit, E. Active Immunization Against hIAPP Oligomers Ameliorates the Diabetes- Associated Phenotype in a Transgenic Mice Model. Sci. Rep. 2017, 7, 14031. [Google Scholar] [CrossRef] [PubMed]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta (BBA) 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Fu, W.; Patel, A.; Kimura, R.; Soudy, R.; Jhamandas, J.H. Amylin Receptor: A Potential Therapeutic Target for Alzheimer’s Disease. Trends Mol. Med. 2017, 23, 709–720. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevcuka, A.; White, K.; Terry, C. Factors That Contribute to hIAPP Amyloidosis in Type 2 Diabetes Mellitus. Life 2022, 12, 583. https://doi.org/10.3390/life12040583
Sevcuka A, White K, Terry C. Factors That Contribute to hIAPP Amyloidosis in Type 2 Diabetes Mellitus. Life. 2022; 12(4):583. https://doi.org/10.3390/life12040583
Chicago/Turabian StyleSevcuka, Adriana, Kenneth White, and Cassandra Terry. 2022. "Factors That Contribute to hIAPP Amyloidosis in Type 2 Diabetes Mellitus" Life 12, no. 4: 583. https://doi.org/10.3390/life12040583
APA StyleSevcuka, A., White, K., & Terry, C. (2022). Factors That Contribute to hIAPP Amyloidosis in Type 2 Diabetes Mellitus. Life, 12(4), 583. https://doi.org/10.3390/life12040583