
α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Systemic Sclerosis
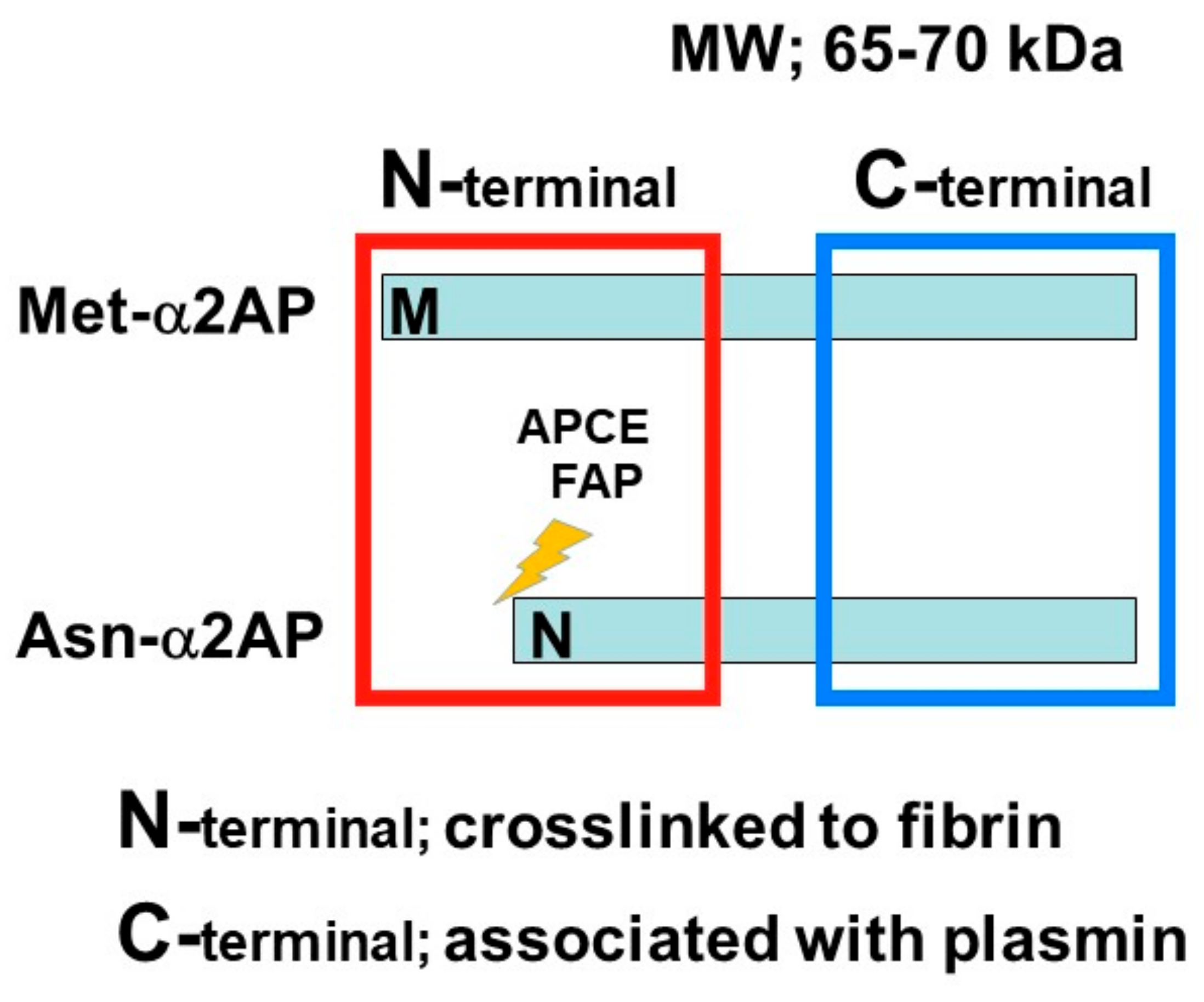
3. α2AP
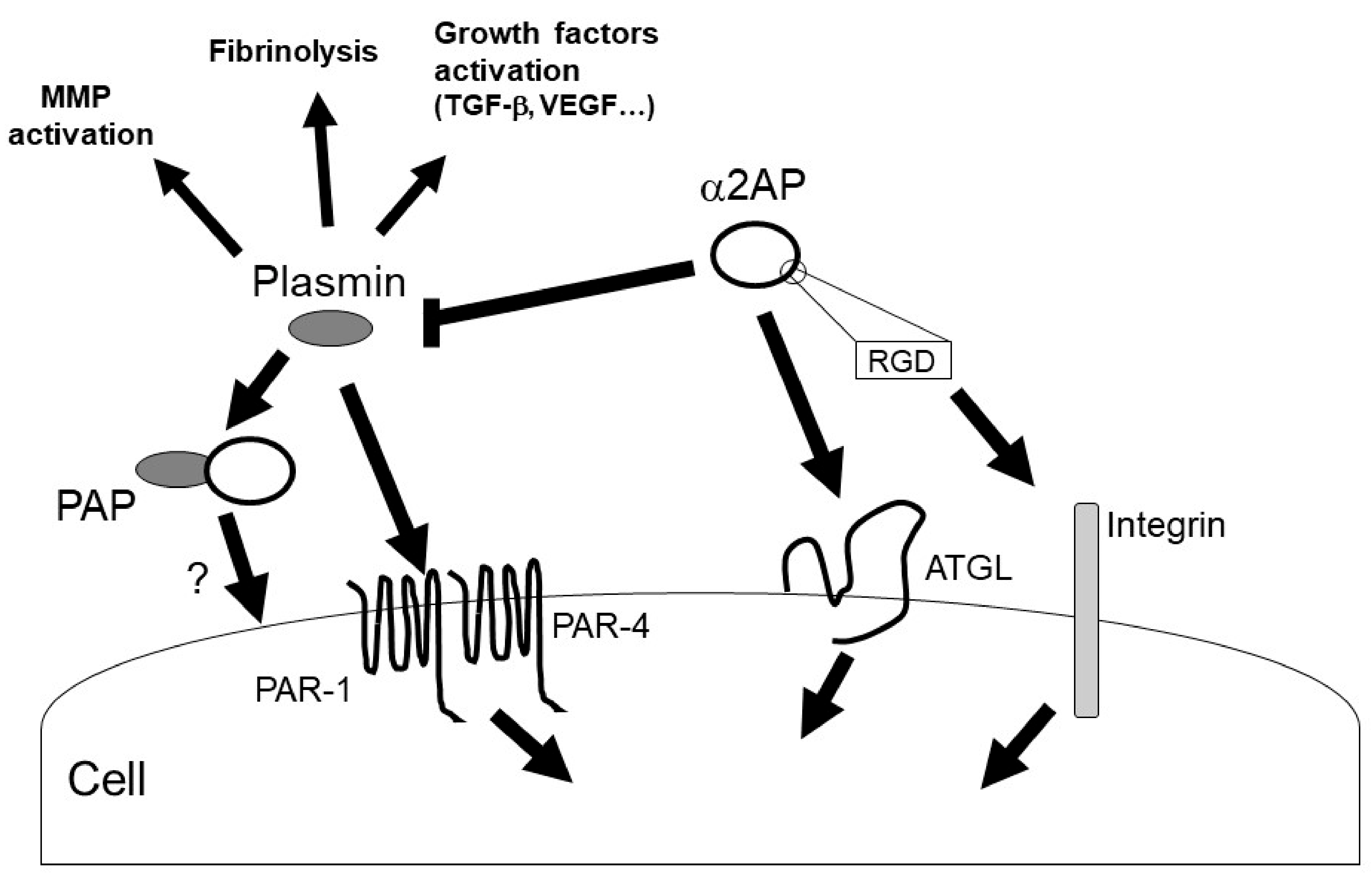
4. α2AP Intracellular Signaling
5. α2AP Deposition in SSc
6. α2AP and Immune Abnormalities in SSc
7. α2AP and Vascular Damage in SSc
8. α2AP and Fibrosis in SSc
9. α2AP and Coagulation/Fibrinolysis in SSc
10. The Role of α2AP as a Therapeutic Target for SSc
11. Conclusions and Therapeutic Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gilbane, A.; Denton, C.; Holmes, A. Scleroderma Pathogenesis: A Pivotal Role for Fibroblasts as Effector Cells. Arthritis Res. 2013, 15, 215. [Google Scholar] [CrossRef] [PubMed]
- Collen, D. Identification and Some Properties of a New Fast-Reacting Plasmin Inhibitor in Human Plasma. Eur. J. Biochem. 1976, 69, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Kaneiwa, A.; Minamida, M.; Kanno, M.; Tomogane, K.; Takeuchi, K.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The Absence of uPAR Is Associated with the Progression of Dermal Fibrosis. J. Investig. Dermatol. 2008, 128, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-Mesenchymal Transition Contributes to Endothelial Dysfunction and Dermal Fibrosis in Systemic Sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef]
- Kanno, Y.; Kuroki, A.; Okada, K.; Tomogane, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-Antiplasmin Is Involved in the Production of Transforming Growth Factor Beta1 and Fibrosis. J. Thromb. Haemost. 2007, 5, 2266–2273. [Google Scholar] [CrossRef]
- Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619. [Google Scholar] [CrossRef]
- Lemaire, R.; Burwell, T.; Sun, H.; Delaney, T.; Bakken, J.; Cheng, L.; Rebelatto, M.C.; Czapiga, M.; De-Mendez, I.; Coyle, A.J.; et al. Resolution of Skin Fibrosis by Neutralization of the Antifibrinolytic Function of Plasminogen Activator Inhibitor 1. Arthritis Rheumatol. 2015, 68, 473–483. [Google Scholar] [CrossRef]
- Lijnen, H.R.; De Cock, F.; Van Hoef, B.; Schlott, B.; Collen, D. Characterization of the Interaction between Plasminogen and Staphylokinase. JBIC J. Biol. Inorg. Chem. 1994, 224, 143–149. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Kuretake, H.; Ikeda, K.; Matsuo, O. uPA Attenuated LPS-Induced Inflammatory Osteoclastogenesis through the Plasmin/PAR-1/Ca2+/CaMKK/AMPK Axis. Int. J. Biol. Sci. 2016, 12, 63–71. [Google Scholar] [CrossRef]
- Kanno, Y.; Hirade, K.; Ishisaki, A.; Nakajima, K.; Suga, H.; Into, T.; Matsushita, K.; Okada, K.; Matsuo, O.; Matsuno, H. Lack of Alpha2-Antiplasmin Improves Cutaneous Wound Healing via Over-Released Vascular Endothelial Growth Factor-Induced Angiogenesis in Wound Lesions. J. Thromb. Haemost. 2006, 4, 1602–1610. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Minamida, M.; Kaneiwa, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. α2-Antiplasmin Is Associated with the Progression of Fibrosis. Am. J. Pathol. 2010, 176, 238–245. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kuretake, H.; Maruyama, C.; Matsuda, A.; Matsuo, O. α2-Antiplasmin Modulates Bone Formation by Negatively Regulating Osteoblast Differentiation and Function. Int. J. Mol. Med. 2017, 40, 854–858. [Google Scholar] [CrossRef]
- Kawashita, E.; Kanno, Y.; Asayama, H.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Involvement of α2-Antiplasmin in Dendritic Growth of Hippocampal Neurons. J. Neurochem. 2013, 126, 58–69. [Google Scholar] [CrossRef]
- Kawashita, E.; Kanno, Y.; Ikeda, K.; Kuretake, H.; Matsuo, O.; Matsuno, H. Altered Behavior in Mice with Deletion of the Alpha2-Antiplasmin Gene. PLoS ONE 2014, 9, e97947. [Google Scholar] [CrossRef]
- Hou, Y.; Okada, K.; Okamoto, C.; Ueshima, S.; Matsuo, O. Alpha2-Antiplasmin Is a Critical Regulator of Angiotensin II–Mediated Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2008, 28, 1257–1262. [Google Scholar] [CrossRef]
- Kager, L.M.; Weehuizen, T.A.; Wiersinga, W.J.; Roelofs, J.J.T.H.; Meijers, J.C.M.; Dondorp, A.M.; Veer, C.V.; Van Der Poll, T. Endogenous α2-Antiplasmin Is Protective during Severe Gram-Negative Sepsis (Melioidosis). Am. J. Respir. Crit. Care Med. 2013, 188, 967–975. [Google Scholar] [CrossRef]
- Kanno, Y.; Tsuchida, K.; Maruyama, C.; Hori, K.; Teramura, H.; Asahi, S.; Matsuo, O.; Ozaki, K.-I. Alpha2-Antiplasmin Deficiency Affects Depression and Anxiety-Like Behavior and Apoptosis Induced by Stress in Mice. J. Basic Clin. Physiol. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Kanoh, H.; Seishima, M. The Antifibrotic Effect of α2AP Neutralization in Systemic Sclerosis Dermal Fibroblasts and Mouse Models of Systemic Sclerosis. J. Investig. Dermatol. 2015, 136, 762–769. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Kanoh, H.; Matsuda, A.; Seishima, M. α2AP Regulates Vascular Alteration by Inhibiting VEGF Signaling in Systemic Sclerosis: The Roles of α2AP in Vascular Dysfunction in Systemic Sclerosis. Arthritis Res. Ther. 2017, 19, 22. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The Role of Endothelial Cells in the Vasculopathy of Systemic Sclerosis: A Systematic Review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef]
- Kavian, N.; Batteux, F. Macro- and Microvascular Disease in Systemic Sclerosis. Vasc. Pharmacol. 2015, 71, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Menoud, P.A.; Sappino, N.; Boudal-Khoshbeen, M.; Vassalli, J.D.; Sappino, A.P. The Kidney Is a Major Site of α(2)-Antiplasmin Production. J. Clin. Investig. 1996, 97, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Pechlivani, N.; Kearney, K.J.; Ajjan, R.A. Fibrinogen and Antifibrinolytic Proteins: Interactions and Future Therapeutics. Int. J. Mol. Sci. 2021, 22, 12537. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Mathew, P. α2-Antiplasmin and Its Deficiency: Fibrinolysis Out of Balance. Haemophilia 2008, 14, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Houng, A.K.; Reed, G.L. Venous Stasis-Induced Fibrinolysis Prevents Thrombosis in Mice: Role of α2-Antiplasmin. Blood 2019, 134, 970–978. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Lee, C.S.; Chun, J.-G.; McKee, P.A. Antiplasmin-Cleaving Enzyme Is a Soluble Form of Fibroblast Activation Protein. Blood 2006, 107, 1397–1404. [Google Scholar] [CrossRef]
- Christiansen, V.J.; Jackson, K.W.; Lee, K.N.; McKee, P.A. Effect of Fibroblast Activation Protein and α2-Antiplasmin Cleaving Enzyme on Collagen Types I, III, and IV. Arch. Biochem. Biophys. 2007, 457, 177–186. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Chung, K.H.; McKee, P.A. A Novel Plasma Proteinase Potentiates α2-Antiplasmin Inhibition of Fibrin Digestion. Blood 2004, 103, 3783–3788. [Google Scholar] [CrossRef]
- Lijnen, H.; Van Hoef, B.; Collen, D. Inactivation of the Serpin α2-Antiplasmin by Stromelysin-1. Biochim. Biophys. Acta-Protein Struct. Mol. Enzym. 2001, 1547, 206–213. [Google Scholar] [CrossRef]
- Irving, J.; Pike, R.; Lesk, A.; Whisstock, J. Phylogeny of the Serpin Superfamily: Implications of Patterns of Amino Acid Conservation for Structure and Function. Genome Res. 2000, 10, 1845–1864. [Google Scholar] [CrossRef]
- Law, R.; Abu-Ssaydeh, D.; Whisstock, J. New Insights into the Structure and Function of the Plasminogen/Plasmin System. Curr. Opin. Struct. Biol. 2013, 23, 836–841. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Aparicio, S.; Xu, X.; Tink, A.R.; Lara, N.; Sawant, S.; Barnstable, C.J.; Zhang, S.S.-M. PEDF and the Serpins: Phylogeny, Sequence Conservation, and Functional Domains. J. Struct. Biol. 2005, 151, 130–150. [Google Scholar] [CrossRef]
- Notari, L.; Baladron, V.; Aroca-Aguilar, J.D.; Balko, N.; Heredia, R.; Meyer, C.; Notario, P.M.; Saravanamuthu, S.; Nueda, M.-L.; Sanchez-Sanchez, F.; et al. Identification of a Lipase-Linked Cell Membrane Receptor for Pigment Epithelium-Derived Factor. J. Biol. Chem. 2006, 281, 38022–38037. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-Antiplasmin Regulates the Development of Dermal Fibrosis in Mice by Prostaglandin F2α Synthesis through Adipose Triglyceride Lipase/Calcium-Independent Phospholipase A2. Arthritis Rheum. 2013, 65, 492–502. [Google Scholar] [CrossRef]
- Kanno, Y.; Kawashita, E.; Kokado, A.; Kuretake, H.; Ikeda, K.; Okada, K.; Seishima, M.; Ueshima, S.; Matsuo, O.; Matsuno, H. α2AP Mediated Myofibroblast Formation and the Development of Renal Fibrosis in Unilateral Ureteral Obstruction. Sci. Rep. 2014, 4, srep05967. [Google Scholar] [CrossRef]
- Ma, S.; Wang, S.; Li, M.; Zhang, Y.; Zhu, P. The Effects of Pigment Epithelium-Derived Factor on Atherosclerosis: Putative Mechanisms of the Process. Lipids Health Dis. 2018, 17, 240. [Google Scholar] [CrossRef]
- Abdul, S.; Leebeek, F.W.G.; Rijken, D.C.; De Willige, S.U. Natural Heterogeneity of α2-Antiplasmin: Functional and Clinical Consequences. Blood 2016, 127, 538–545. [Google Scholar] [CrossRef]
- Kanno, Y.; Miyashita, M.; Seishima, M.; Matsuo, O. α2AP Is Associated with the Development of Lupus Nephritis through the Regulation of Plasmin Inhibition and Inflammatory Responses. Immun. Inflamm. Dis. 2020, 8, 267–278. [Google Scholar] [CrossRef]
- Shiomi, A.; Kawao, N.; Yano, M.; Okada, K.; Tamura, Y.; Okumoto, K.; Matsuo, O.; Akagi, M.; Kaji, H. α2-Antiplasmin Is Involved in Bone Loss Induced by Ovariectomy in Mice. Bone 2015, 79, 233–241. [Google Scholar] [CrossRef]
- Kanno, Y.; Hirota, M.; Matsuo, O.; Ozaki, K.-I. α2-Antiplasmin Positively Regulates Endothelial-to-Mesenchymal Transition and Fibrosis Progression in Diabetic Nephropathy. Mol. Biol. Rep. 2021, 49, 205–215. [Google Scholar] [CrossRef]
- Thomas, L.; Moore, N.R.; Miller, S.; Booth, N.A. The C-Terminus of α2-Antiplasmin Interacts with Endothelial Cells. Br. J. Haematol. 2006, 136, 472–479. [Google Scholar] [CrossRef]
- Udvardy, M.; Schwartzott, D.; Jackson, K.; McKee, P. Hybrid Peptide Containing RGDF (Arg-Gly-Asp-Phe) Coupled with the Carboxy Terminal Part of Alpha 2-Antiplasmin Capable of Inhibiting Platelet Aggregation and Promoting Fibrinolysis. Blood Coagul. Fibrinolysis 1995, 6, 11–16. [Google Scholar] [CrossRef]
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Chosa, N.; Nakajima, K.; Nishihara, T.; Toyoshima, K.; Okada, K.; Ueshima, S.; Matsushita, K.; et al. Plasminogen/Plasmin Modulates Bone Metabolism by Regulating the Osteoblast and Osteoclast Function. J. Biol. Chem. 2011, 286, 8952–8960. [Google Scholar] [CrossRef]
- Syrovets, T.; Lunov, O.; Simmet, T. Plasmin as a Proinflammatory Cell Activator. J. Leukoc. Biol. 2012, 92, 509–519. [Google Scholar] [CrossRef]
- Li, X.; Syrovets, T.; Genze, F.; Pitterle, K.; Oberhuber, A.; Orend, K.-H.; Simmet, T. Plasmin Triggers Chemotaxis of Monocyte-Derived Dendritic Cells through an Akt2-Dependent Pathway and Promotes a T-Helper Type-1 Response. Arter. Thromb. Vasc. Biol. 2010, 30, 582–590. [Google Scholar] [CrossRef]
- Draxler, D.F.; Sashindranath, M.; Medcalf, R.L. Plasmin: A Modulator of Immune Function. Semin. Thromb. Hemost. 2017, 43, 143–153. [Google Scholar] [CrossRef]
- Hattori, N.; Mizuno, S.; Yoshida, Y.; Chin, K.; Mishima, M.; Sisson, T.H.; Simon, R.H.; Nakamura, T.; Miyake, M. The Plasminogen Activation System Reduces Fibrosis in the Lung by a Hepatocyte Growth Factor-Dependent Mechanism. Am. J. Pathol. 2004, 164, 1091–1098. [Google Scholar] [CrossRef]
- Kuliopulos, A.; Covic, L.; Seeley, S.K.; Sheridan, P.J.; Helin, J.; Costello, C.E. Plasmin Desensitization of the PAR1 Thrombin Receptor: Kinetics, Sites of Truncation, and Implications for Thrombolytic Therapy. Biochemistry 1999, 38, 4572–4585. [Google Scholar] [CrossRef]
- Quinton, T.M.; Kim, S.; Derian, C.K.; Jin, J.; Kunapuli, S.P.; Reynolds, L.F.; de Bettignies, C.; Norton, T.; Beeser, A.; Chernoff, J.; et al. Plasmin-Mediated Activation of Platelets Occurs by Cleavage of Protease-Activated Receptor 4. J. Biol. Chem. 2004, 279, 18434–18439. [Google Scholar] [CrossRef]
- Trejo, J.; Stover, T.; Kester, M. Protease-Activated Receptors: New Concepts in Regulation of G Protein-Coupled Receptor Signaling and Trafficking. J. Pharmacol. Exp. Ther. 2003, 307, 437–442. [Google Scholar] [CrossRef]
- Jinnin, M.; Ihn, H.; Yamane, K.; Asano, Y.; Yazawa, N.; Tamaki, K. Plasma Plasmin-Alpha2-Plasmin Inhibitor Complex Levels Are Increased in Systemic Sclerosis Patients with Pulmonary Hypertension. Rheumatology 2003, 42, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Bălănescu, P.; Lădaru, A.; Bălănescu, E.; Nicolau, A.; Băicuş, C.; Dan, G. IL-17, IL-6 and IFN-γ in Systemic Sclerosis Patients. Rom. J. Intern. Med. 2015, 53, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, A.; Komura, K.; Iwata, Y.; Ogawa, F.; Hara, T.; Muroi, E.; Takenaka, M.; Shimizu, K.; Hasegawa, M.; Fujimoto, M.; et al. Clinical Significance of Serum HMGB-1 and sRAGE Levels in Systemic Sclerosis: Association with Disease Severity. J. Clin. Immunol. 2008, 29, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-Connective Tissue Growth Factor (CTGF/CCN2) Monoclonal Antibody Attenuates Skin Fibrosis in Mice Models of Systemic Sclerosis. Arthritis Res. Ther. 2017, 19, 134. [Google Scholar] [CrossRef]
- Yamashita, T.; Asano, Y.; Taniguchi, T.; Nakamura, K.; Saigusa, R.; Miura, S.; Toyama, T.; Takahashi, T.; Ichimura, Y.; Yoshizaki, A.; et al. Glycyrrhizin Ameliorates Fibrosis, Vasculopathy, and Inflammation in Animal Models of Systemic Sclerosis. J. Investig. Dermatol. 2016, 137, 631–640. [Google Scholar] [CrossRef]
- Niwa, H.; Kanno, Y.; Shu, E.; Seishima, M. Decrease in Matrix Metalloproteinase-3 Activity in Systemic Sclerosis Fibroblasts Causes α2-Antiplasmin and Extracellular Matrix Deposition, and Contributes to Fibrosis Development. Mol. Med. Rep. 2020, 22, 3001–3007. [Google Scholar] [CrossRef]
- Young-Min, S.A.; Beeton, C.; Laughton, R.; Plumpton, T.; Bartram, S.; Murphy, G.; Black, C.; Cawston, T.E. Serum TIMP-1, TIMP-2, and MMP-1 in Patients with Systemic Sclerosis, Primary Raynaud’s Phenomenon, and in Normal Controls. Ann. Rheum. Dis. 2001, 60, 846–851. [Google Scholar]
- Nishijima, C.; Hayakawa, I.; Matsushita, T.; Komura, K.; Hasegawa, M.; Takehara, K.; Sato, S. Autoantibody against Matrix Metalloproteinase-3 in Patients with Systemic Sclerosis. Clin. Exp. Immunol. 2004, 138, 357–363. [Google Scholar] [CrossRef]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of Monocyte/Macrophage Subsets in the Skin and Peripheral Blood Derived from Patients with Systemic Sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef]
- O’Reilly, S.; Hügle, T.; Van Laar, J.M. T Cells in Systemic Sclerosis: A Reappraisal. Rheumatology 2012, 51, 1540–1549. [Google Scholar] [CrossRef]
- Sanges, S.; Guerrier, T.; Launay, D.; Lefèvre, G.; Labalette, M.; Forestier, A.; Sobanski, V.; Corli, J.; Hauspie, C.; Jendoubi, M.; et al. Role of B Cells in the Pathogenesis of Systemic Sclerosis. Rev. Médecine Interne 2016, 38, 113–124. [Google Scholar] [CrossRef]
- Fuschiotti, P. Current Perspectives on the Immunopathogenesis of Systemic Sclerosis. ImmunoTargets Ther. 2016, 5, 21–35. [Google Scholar] [CrossRef]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.; Postlethwaite, A. Pathogenesis of Systemic Sclerosis. Front. Immunol. 2015, 6, 272. [Google Scholar] [CrossRef]
- Brown, M.; O’Reilly, S. The Immunopathogenesis of Fibrosis in Systemic Sclerosis. Clin. Exp. Immunol. 2018, 195, 310–321. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nishioka, K. Animal Model of Sclerotic Skin. IV: Induction of Dermal Sclerosis by Bleomycin is T Cell Independent. J. Investig. Dermatol. 2001, 117, 999–1001. [Google Scholar] [CrossRef][Green Version]
- Kanno, Y.; Shu, E.; Niwa, H.; Kanoh, H.; Seishima, M. Alternatively Activated Macrophages Are Associated with the α2AP Production that Occurs with the Development of Dermal Fibrosis: The Role of Alternatively Activated Macrophages on the Development of Fibrosis. Arthritis Res. Ther. 2020, 22, 76. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C. Macrophages: Friend or Foe in Idiopathic Pulmonary Fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Nakayama, W.; Jinnin, M.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Ihn, H. Serum Levels of Soluble CD163 in Patients with Systemic Sclerosis. Rheumatol. Int. 2010, 32, 403–407. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Tsukui, T.; Shichino, S.; Shimaoka, T.; Ueha, S.; Matsushima, K. Cellular and Molecular Mechanisms of Chronic Inflammation-Associated Organ Fibrosis. In Chronic Inflammation; Springer: Tokyo, Japan, 2016; pp. 19–36. [Google Scholar] [CrossRef]
- Nunes, J.P.L.; Cunha, A.C.; Meirinhos, T.; Nunes, A.; Araújo, P.M.; Godinho, A.R.; Vilela, E.M.; Vaz, C. Prevalence of Auto-Antibodies Associated to Pulmonary Arterial Hypertension in Scleroderma—A Review. Autoimmun. Rev. 2018, 17, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; Huigens, C.A.; Hügle, T.; Stanly, T.; Gessner, A.; Griffiths, B.; Radstake, T.R.D.J.; Hambleton, S.; O’Reilly, S.; Van Laar, J.M. Toll-Like Receptor-Mediated, Enhanced Production of Profibrotic TIMP-1 in Monocytes from Patients with Systemic Sclerosis: Role of Serum Factors. Ann. Rheum. Dis. 2013, 72, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, N.; Kikuchi, K.; Ihn, H.; Fujimoto, M.; Kubo, M.; Tamaki, T.; Tamaki, K. Serum Levels of Tissue Inhibitor of MetalloProteinases 2 in Patients with Systemic Sclerosis. J. Am. Acad. Dermatol. 2000, 42, 70–75. [Google Scholar] [CrossRef]
- Elias, G.J.; Ioannis, M.; Theodora, P.; Dimitrios, P.P.; Despoina, P.; Kostantinos, V.; Charalampos, K.; Vassilios, V.; Petros, S.P. Circulating Tissue Inhibitor of Matrix Metalloproteinase-4 (TIMP-4) in Systemic Sclerosis Patients with Elevated Pulmonary Arterial Pressure. Mediat. Inflamm. 2008, 2008, 164134. [Google Scholar] [CrossRef] [PubMed]
- Tomimura, S.; Ogawa, F.; Iwata, Y.; Komura, K.; Hara, T.; Muroi, E.; Takenaka, M.; Shimizu, K.; Hasegawa, M.; Fujimoto, M.; et al. Autoantibodies against Matrix Metalloproteinase-1 in Patients with Localized Scleroderma. J. Dermatol. Sci. 2008, 52, 47–54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okada, K.; Ueshima, S.; Kawao, N.; Yano, M.; Tamura, Y.; Tanaka, M.; Sakamoto, A.; Hatano, M.; Arima, M.; Miyata, S.; et al. Lack of Both α2-Antiplasmin and Plasminogen Activator Inhibitor Type-1 Induces High IgE Production. Life Sci. 2013, 93, 89–95. [Google Scholar] [CrossRef]
- Eddy, J.L.; Schroeder, J.A.; Zimbler, D.L.; Bellows, L.E.; Lathem, W.W. Impact of the Pla Protease Substrate α2-Antiplasmin on the Progression of Primary Pneumonic Plague. Infect. Immun. 2015, 83, 4837–4847. [Google Scholar] [CrossRef]
- Zhabin, S.G.; Gorin, V.S. The Effects of Alpha 2-Antiplasmin Complex and Alpha 2-Antiplasmin on the Secretion of IgG and IgM by Cultured Human Mononuclear Cells. J. Clin. Lab. Immunol. 1997, 49, 77–82. [Google Scholar]
- Didiasova, M.; Wujak, L.; Wygrecka, M.; Zakrzewicz, D. From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer. Int. J. Mol. Sci. 2014, 15, 21229–21252. [Google Scholar] [CrossRef]
- Kanno, Y.; Sakai, A.; Miyashita, M.; Tsuchida, K.; Matsuo, O. Plasminogen Deficiency Is Associated with Improved Glucose Tolerance, and Lower DPP-4 Activity. Diabetes Res. Clin. Pr. 2016, 120, 190–193. [Google Scholar] [CrossRef]
- Gomez-Salinero, J.M.; Rafii, S. Plasmin Regulation of Acute Cytokine Storm. Blood 2017, 130, 5–6. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Cell Surface Remodeling by Plasmin: A New Function for an Old Enzyme. J. Biomed. Biotechnol. 2012, 2012, 564259. [Google Scholar] [CrossRef]
- Das, R.; Ganapathy, S.; Settle, M.; Plow, E.F. Plasminogen Promotes Macrophage Phagocytosis in Mice. Blood 2014, 124, 679–688. [Google Scholar] [CrossRef]
- Lorenz, N.; Loef, E.J.; Kelch, I.D.; Verdon, D.J.; Black, M.M.; Middleditch, M.J.; Greenwood, D.R.; Graham, E.S.; Brooks, A.E.; Dunbar, P.R.; et al. Plasmin and Regulators of Plasmin Activity Control the Migratory Capacity and Adhesion of Human T Cells and Dendritic Cells by Regulating Cleavage of the Chemokine CCL21. Immunol. Cell Biol. 2016, 94, 955–963. [Google Scholar] [CrossRef]
- Shimazu, H.; Munakata, S.; Tashiro, Y.; Salama, Y.; Dhahri, D.; Eiamboonsert, S.; Ota, Y.; Onoda, H.; Tsuda, Y.; Okada, Y.; et al. Pharmacological Targeting of Plasmin Prevents Lethality in a Murine Model of Macrophage Activation Syndrome. Blood 2017, 130, 59–72. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular Intercommunication between the Complement and Coagulation Systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef]
- Ward, J.R.; Dower, S.K.; Whyte, M.K.; Buttle, D.J.; Sabroe, I. Potentiation of TLR4 signalling by Plasmin Activity. Biochem. Biophys. Res. Commun. 2006, 341, 299–303. [Google Scholar] [CrossRef]
- Block, J.; Sequeira, W. Raynaud’s Phenomenon. Lancet 2001, 357, 2042–2048. [Google Scholar] [CrossRef]
- Walker, J.G.; Stirling, J.; Beroukas, D.; Dharmapatni, K.; Haynes, D.R.; Smith, M.D.; Ahern, M.J.; Roberts-Thomson, P.J. Histopathological and Ultrastructural Features of Dermal Telangiectasias in Systemic Sclerosis. Pathology 2005, 37, 220–225. [Google Scholar] [CrossRef]
- Au, K.; Singh, M.K.; Bodukam, V.K.; Bae, S.; Maranian, P.; Ogawa, R.; Spiegel, B.; McMahon, M.; Hahn, B.; Khanna, D. Atherosclerosis in Systemic Sclerosis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2011, 63, 2078–2090. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Mechanisms in the Loss of Capillaries in Systemic Sclerosis: Angiogenesis versus Vasculogenesis. J. Cell. Mol. Med. 2010, 14, 1241–1254. [Google Scholar] [CrossRef]
- Liu, X.; Gorzelanny, C.; Schneider, S.W. Platelets in Skin Autoimmune Diseases. Front. Immunol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Aloui, C.; Prigent, A.; Tariket, S.; Sut, C.; Fagan, J.; Cognasse, F.; Chakroun, T.; Garraud, O.; Laradi, S. Levels of Human Platelet-Derived Soluble CD40 Ligand Depend on Haplotypes of CD40LG-CD40-ITGA2. Sci. Rep. 2016, 6, 24715. [Google Scholar] [CrossRef]
- Liakouli, V.; Cipriani, P.; Marrelli, A.; Alvaro, S.; Ruscitti, P.; Giacomelli, R. Angiogenic Cytokines and Growth Factors in Systemic Sclerosis. Autoimmun. Rev. 2011, 10, 590–594. [Google Scholar] [CrossRef]
- Trojanowska, M. Cellular and Molecular Aspects of Vascular Dysfunction in Systemic Sclerosis. Nat. Rev. Rheumatol. 2010, 6, 453–460. [Google Scholar] [CrossRef]
- Zaman, M.; Oparil, S.; Calhoun, D. Drugs Targeting the Renin-Angiotensin-Aldosterone System. Nat. Rev. Drug Discov. 2002, 1, 621–636. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Takagi, K.; Hara, M.; Fukasawa, C.; Sugiura, T.; Nishimagi, E.; Harigai, M.; Kamatani, N. Angiotensin II in the lesional Skin of Systemic Sclerosis Patients Contributes to Tissue Fibrosis via Angiotensin II Type 1 Receptors. Arthritis Care Res. 2004, 50, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Dadoniene, J.; Cypiene, A.; Ryliskyte, L.; Rugiene, R.; Ryliškiene, K.; Laucevičius, A. Skin Autofluorescence in Systemic Sclerosis Is Related to the Disease and Vascular Damage: A Cross-Sectional Analytic Study of Comparative Groups. Dis. Markers. 2015, 2015, 837470. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.A.; Herrick, A.L.; Cordingley, L.; Freemont, A.J.; Jeziorska, M. Expression of Advanced Glycation end Products and Their Receptor in Skin from Patients with Systemic Sclerosis with and without Calcinosis. Rheumatology 2009, 48, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yao, Y.-C.; Gu, X.-Q.; Che, D.; Ma, C.-Q.; Dai, Z.; Li, C.; Zhou, T.; Cai, W.-B.; Yang, Z.-H.; et al. Plasminogen Kringle 5 Induces Endothelial Cell Apoptosis by Triggering a Voltage-dependent Anion Channel 1 (VDAC1) Positive Feedback Loop. J. Biol. Chem. 2014, 289, 32628–32638. [Google Scholar] [CrossRef] [PubMed]
- Okajima, K.; Abe, H.; Binder, B.R. Endothelial Cell Injury Induced by Plasmin in Vitro. J. Lab. Clin. Med. 1995, 126, 377–384. [Google Scholar]
- Mosesson, M.W. Fibrinogen and Fibrin Structure and Functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Watabe, A.; Ohta, M.; Matsuyama, N.; Mizuno, K.; El Borai, N.; Tanimoto, T.; Kawanishi, T.; Hayakawa, T. Characterization of Plasmin-Induced Platelet Aggregation. Res. Commun. Mol. Pathol. Pharmacol. 1997, 96, 341–352. [Google Scholar]
- Niewiarowski, S.; Senyi, A.F.; Gillies, P. Plasmin-Induced Platelet Aggregation and Platelet Release Reaction. Effects on Hemostasis. J. Clin. Investig. 1973, 52, 1647–1659. [Google Scholar] [CrossRef]
- Rundhaug, J.E. Matrix Metalloproteinases and Angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Yan, Q.; Sage, E. Transforming Growth Factor-Beta1 Induces Apoptotic Cell Death in Cultured Retinal Endothelial Cells but not Pericytes: Association with Decreased Expression of p21waf1/cip1. J. Cell Biochem. 1998, 70, 70–83. [Google Scholar] [CrossRef]
- Gamal, R.M.; Gamal, W.M.; Abozaid, H.S.M.; Ghandour, A.M.; Mohamed, M.E.; Emad, Y.; Galeel, A.A. 201 Study of the Osteoprotegerin Receptor Activator of Nuclear Factor kB Ligand System Association with Inflammation and Atherosclerosis in Systemic Sclerosis. Rheumatology 2018, 57, key075-425. [Google Scholar] [CrossRef]
- Van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Xing, X.; Li, A.; Tan, H.; Zhou, Y. IFN-γ+ IL-17+ Th17 Cells Regulate Fibrosis through Secreting IL-21 in Systemic Scleroderma. J. Cell Mol. Med. 2020, 24, 13600–13608. [Google Scholar] [CrossRef]
- Vernon, M.A.; Mylonas, K.J.; Hughes, J. Macrophages and Renal Fibrosis. Semin. Nephrol. 2010, 30, 302–317. [Google Scholar] [CrossRef]
- Cracowski, J.; Marpeau, C.; Carpentier, P.; Imbert, B.; Hunt, M.; Stanke-Labesque, F.; Bessard, G. Enhanced in Vivo Lipid Peroxidation in Scleroderma Spectrum Disorders. Arthritis Rheum. 2001, 44, 1143–1148. [Google Scholar] [CrossRef]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F2alpha Receptor Signaling Facilitates Bleomycin-Induced Pulmonary Fibrosis Independently of Transforming Growth Factor-Beta. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef]
- Vona, R.; Giovannetti, A.; Gambardella, L.; Malorni, W.; Pietraforte, D.; Straface, E. Oxidative Stress in the Pathogenesis of Systemic Scleroderma: An Overview. J. Cell. Mol. Med. 2018, 22, 3308–3314. [Google Scholar] [CrossRef]
- Mancini, O.K.; Acevedo, M.; Fazez, N.; Cuillerier, A.; Ruiz, A.F.; Huynh, D.N.; Burelle, Y.; Ferbeyre, G.; Baron, M.; Servant, M.J. Oxidative Stress-Induced Senescence Mediates Inflammatory and Fibrotic Phenotypes in Fibroblasts from Systemic Sclerosis Patients. Rheumatology 2022, 61, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Thuan, D.T.B.; Zayed, H.; Eid, A.H.; Abou-Saleh, H.; Nasrallah, G.; Mangoni, A.A.; Pintus, G. A Potential Link between Oxidative Stress and Endothelial-to-Mesenchymal Transition in Systemic Sclerosis. Front. Immunol. 2018, 9, 1985. [Google Scholar] [CrossRef]
- Kawashita, E.; Ishihara, K.; Miyaji, H.; Tanishima, Y.; Kiriyama, A.; Matsuo, O.; Akiba, S. α2-Antiplasmin as a Potential Regulator of the Spatial Memory Process and Age-Related Cognitive Decline. Mol. Brain 2020, 13, 140. [Google Scholar] [CrossRef]
- Bobik, A.; Tkachuk, V. Metalloproteinases and Plasminogen Activators in Vessel Remodeling. Curr. Hypertens. Rep. 2003, 5, 466–472. [Google Scholar] [CrossRef]
- Waasdorp, M.; Duitman, J.; Spek, C.A. Plasmin Reduces Fibronectin Deposition by Mesangial Cells in a Protease-Activated Receptor-1 Independent Manner. Biochem. Biophys. Rep. 2017, 10, 152–156. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Rogers, D.S.; Simon, R.H.; Sisson, T.H.; Thannickal, V.J. Plasminogen Activation–Induced Pericellular Fibronectin Proteolysis Promotes Fibroblast Apoptosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 78–87. [Google Scholar] [CrossRef]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; et al. The Antifibrotic Effects of Plasminogen Activation Occur via Prostaglandin E2 Synthesis in Humans and Mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef]
- Kochtebane, N.; Choqueux, C.; Passefort, S.; Nataf, P.; Messika-Zeitoun, D.; Bartagi, A.; Michel, J.-B.; Anglés-Cano, E.; Jacob, M.-P. Plasmin Induces Apoptosis of Aortic Valvular Myofibroblasts. J. Pathol. 2009, 221, 37–48. [Google Scholar] [CrossRef]
- Asano, Y. Systemic Sclerosis. J. Dermatol. 2018, 45, 128–138. [Google Scholar] [CrossRef]
- Cerinic, M.M.; Valentini, G.; Sorano, G.; D’Angelo, S.; Cuomo, G.; Fenu, L.; Generini, S.; Cinotti, S.; Morfini, M.; Pignone, A.; et al. Blood Coagulation, Fibrinolysis, and Markers of Endothelial Dysfunction in Systemic Sclerosis. Semin. Arthritis Rheum. 2003, 32, 285–295. [Google Scholar] [CrossRef]
- Terrier, B.; Tamby, M.; Camoin, L.; Guilpain, P.; Bérezné, A.; Tamas, N.; Broussard, C.; Hotellier, F.; Humbert, M.; Simonneau, G.; et al. Antifibroblast Antibodies from Systemic Sclerosis Patients Bind to {Alpha}-Enolase and Are Associated with Interstitial Lung Disease. Ann. Rheum. Dis. 2010, 69, 428–433. [Google Scholar] [CrossRef]
- Ntelis, K.; Solomou, E.E.; Sakkas, L.; Liossis, S.-N.; Daoussis, D. The Role of Platelets in Autoimmunity, Vasculopathy, and Fibrosis: Implications for Systemic Sclerosis. Semin. Arthritis Rheum. 2017, 47, 409–417. [Google Scholar] [CrossRef]
- De Giorgio-Miller, A.; Bottoms, S.; Laurent, G.; Carmeliet, P.; Herrick, S. Fibrin-Induced Skin Fibrosis in Mice Deficient in Tissue Plasminogen Activator. Am. J. Pathol. 2005, 167, 721–732. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Schoenecker, J.G.; Flick, M.J. The Multifaceted Role of Fibrinogen in Tissue Injury and Inflammation. Blood 2019, 133, 511–520. [Google Scholar] [CrossRef]
- Schuliga, M. The Inflammatory Actions of Coagulant and Fibrinolytic Proteases in Disease. Mediat. Inflamm. 2015, 2015, 437695. [Google Scholar] [CrossRef]
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The Fibrogenic Actions of the Coagulant and Plasminogen Activation Systems in Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2018, 97, 108–117. [Google Scholar] [CrossRef]
- Miniati, M.; Fiorillo, C.; Becatti, M.; Monti, S.; Bottai, M.; Marini, C.; Grifoni, E.; Formichi, B.; Bauleo, C.; Arcangeli, C.; et al. Fibrin Resistance to Lysis in Patients with Pulmonary Hypertension Other than Thromboembolic. Am. J. Respir. Crit. Care Med. 2010, 181, 992–996. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a Key Regulator of Inflammation in Disease. Semin. Immunopathol. 2011, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Rubel, C.; Fernández, G.C.; Dran, G.; Bompadre, M.B.; Isturiz, M.A.; Palermo, M.S. Fibrinogen Promotes Neutrophil Activation and Delays Apoptosis. J. Immunol. 2001, 166, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, R.G.; Pan, P.M.; Srikanth, S.; Todd, R.F. Fibrinogen Activates NF-Kappa B Transcription Factors in Mononuclear Phagocytes. J. Immunol. 1998, 161, 1462–1470. [Google Scholar] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen Stimulates Macrophage Chemokine Secretion Through Toll-Like Receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef]
- Suehiro, K.; Gailit, J.; Plow, E. Fibrinogen Is a Ligand for Integrin α5β1 on Endothelial Cells. J. Biol. Chem. 1997, 272, 5360–5366. [Google Scholar] [CrossRef]
- Yokoyama, K.; Zhang, X.; Medved, L.; Takada, Y. Specific Binding of Integrin α vs. β3 to the Fibrinogen γ and αE Chain C-Terminal Domains. Biochemistry 1999, 38, 5872–5877. [Google Scholar] [CrossRef]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.-Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of Fibrinogen by Proteinases Elicits Allergic Responses Through Toll-Like Receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef]
- Yakovlev, S.; Mikhailenko, I.; Cao, C.; Zhang, L.; Strickland, D.K.; Medved, L. Identification of VLDLR as a Novel Endothelial Cell Receptor for Fibrin that Modulates Fibrin-Dependent Transendothelial Migration of Leukocytes. Blood 2012, 119, 637–644. [Google Scholar] [CrossRef]
- Sanchez-Pernaute, O.; Filkova, M.; Gabucio, A.; Klein, M.; Maciejewska-Rodrigues, H.; Ospelt, C.; Brentano, F.; Michel, B.A.; Gay, R.E.; Herrero-Beaumont, G.; et al. Citrullination Enhances the Pro-Inflammatory Response to Fibrin in Rheumatoid Arthritis Synovial Fibroblasts. Ann. Rheum. Dis. 2012, 72, 1400–1406. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef]
- Herwald, H.; Korte, W.; Allanore, Y.; Denton, C.P.; Cerinic, M.M.; Dickneite, G. Coagulation Factor XIII: A Multifunctional Transglutaminase with Clinical Potential in a Range of Conditions. Thromb. Haemost. 2015, 113, 686–697. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Wang, H.; Liu, J. Study on the Influence of Oxidative Stress on the Fibrillization of Fibrinogen. Biochem. J. 2016, 473, 4373–4384. [Google Scholar] [CrossRef]
- Siudut, J.; Natorska, J.; Wypasek, E.; Wiewiórka, Ł.; Ostrowska-Kaim, E.; Wiśniowska-Śmiałek, S.; Plens, K.; Legutko, J.; Undas, A. Impaired Fibrinolysis in Patients with Isolated Aortic Stenosis is Associated with Enhanced Oxidative Stress. J. Clin. Med. 2020, 9, 2002. [Google Scholar] [CrossRef]
- Keane, F.; Nadvi, N.; Yao, T.; Gorrell, M. Neuropeptide Y, B-Type Natriuretic Peptide, Substance P and Peptide YY Are Novel Substrates of Fibroblast Activation Protein-α. FEBS J. 2011, 278, 1316–1332. [Google Scholar] [CrossRef]
- Soare, A.; Györfi, H.A.; Matei, A.E.; Dees, C.; Rauber, S.; Wohlfahrt, T.; Chen, C.W.; Ludolph, I.; Horch, R.E.; Bäuerle, T.; et al. Dipeptidyl-Peptidase-4 as a Marker of Activated Fibroblasts and a Potential Target for the Treatment of Fibrosis in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 137–149. [Google Scholar] [CrossRef]
- Reed, G.; Houng, A.; Wang, D. Microvascular Thrombosis, Fibrinolysis, Ischemic Injury, and Death after Cerebral Thromboembolism Are Affected by Levels of Circulating α2-Antiplasmin. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2586–2593. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Dolence, E.K.; Mckee, P.A. Enhancement of Fibrinolysis by Inhibiting Enzymatic Cleavage of Precursor α2-Antiplasmin. J. Thromb. Haemost. 2011, 9, 987–996. [Google Scholar] [CrossRef]
- Szabo, I.; Muntean, L.; Crisan, T.; Rednic, V.; Sirbe, C.; Rednic, S. Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs. Biomedicines 2021, 9, 1471. [Google Scholar] [CrossRef]
- Ciechomska, M.; O’Reilly, S.; Suwara, M.; Bogunia-Kubik, K.; van Laar, J. MiR-29a Reduces TIMP-1 Production by Dermal Fibroblasts via Targeting TGF-β Activated Kinase 1 Binding Protein 1, Implications for Systemic Sclerosis. PLoS ONE 2014, 30, e115596. [Google Scholar] [CrossRef]
- Kanno, Y.; Shu, E.; Niwa, H.; Seishima, M.; Ozaki, K.-I. MicroRNA-30c Attenuates Fibrosis Progression and Vascular Dysfunction in Systemic Sclerosis Model Mice. Mol. Biol. Rep. 2021, 48, 3431–3437. [Google Scholar] [CrossRef]
- Hirman, A.R.; Du, L.; Cheng, S.; Zheng, H.; Duo, L.; Zhai, Q.; Xu, J. MiR-133a-3p Inhibits Scar Formation in Scalded Mice and Suppresses the Proliferation and Migration of Scar Derived-Fibroblasts by Targeting Connective Tissue Growth Factor. Exp. Anim. 2021, 70, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, H.; Wang, B.; Yuan, Y.; Klein, J.D.; Wang, X.H. Exogenous miR-26a Suppresses Muscle Wasting and Renal Fibrosis in Obstructive Kidney Disease. FASEB J. 2019, 33, 13590–13601. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ji, Q.; Zhu, H.; Ren, Y.; Fan, Z.; Tian, N. miR-30a Attenuates Cardiac Fibrosis in Rats with Myocardial Infarction by Inhibiting CTGF. Exp. Ther. Med. 2018, 15, 4318–4324. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, W.P.; Eltringham-Smith, L.J.; Gataiance, S.; Bhakta, V. Addition of a Sequence from α2-Antiplasmin Transforms Human Serum Albumin into a Blood Clot Component that Speeds Clot Lysis. BMC Biotechnol. 2009, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Jackson, K.W.; McKee, P.A. Effect of a Synthetic Carboxy-Terminal Peptide of α2-Antiplasmin on Urokinase-Induced Fibrinolysis. Thromb. Res. 2002, 105, 263–270. [Google Scholar] [CrossRef]
- Shi, G.Y.; Wu, H.L. Isolation and Characterization of Microplasminogen. A Low Molecular Weight Form of Plasminogen. J. Biol. Chem. 1988, 263, 17071–17075. [Google Scholar] [CrossRef]
- Pakola, S.; Cahillane, G.; Stassen, J.; Lijnen, H.; Verhamme, P. Neutralization of α(2)-Antiplasmin by Microplasmin: A Randomized, Double-Blind, Placebo-Controlled, Ascending-Dose Study in Healthy Male Volunteers. Clin. Ther. 2009, 31, 1688–1706. [Google Scholar] [CrossRef]
- Nagai, N.; De Mol, M.; Van Hoef, B.; Verstreken, M.; Collen, D. Depletion of Circulating α(2)-Antiplasmin by Intravenous Plasmin or Immunoneutralization Reduces Focal Cerebral Ischemic Injury in the Absence of Arterial Recanalization. Blood 2001, 97, 3086–3092. [Google Scholar] [CrossRef]
- Suzuki, Y.; Chen, F.; Ni, Y.; Marchal, G.; Collen, D.; Nagai, N. Microplasmin Reduces Ischemic Brain Damage and Improves Neurological Function in a Rat Stroke Model Monitored with MRI. Stroke 2004, 35, 2402–2406. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, Y.; Shu, E. α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis. Life 2022, 12, 396. https://doi.org/10.3390/life12030396
Kanno Y, Shu E. α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis. Life. 2022; 12(3):396. https://doi.org/10.3390/life12030396
Chicago/Turabian StyleKanno, Yosuke, and En Shu. 2022. "α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis" Life 12, no. 3: 396. https://doi.org/10.3390/life12030396
APA StyleKanno, Y., & Shu, E. (2022). α2-Antiplasmin as a Potential Therapeutic Target for Systemic Sclerosis. Life, 12(3), 396. https://doi.org/10.3390/life12030396