
Clays and the Origin of Life: The Experiments


Abstract
1. Introduction
2. Clay Minerals on Early Earth
3. Clay Minerals Found in Our Solar System
3.1. Mars
3.2. Meteorites
3.3. Asteroids and Comets
4. The Search for Life on Mars
“analysis of the results shows that a small but significant formation of organic matter occurred” and that the sterilized control showed no evidence of organics, showing that the “findings could be attributed to biological activity [74]”.
“an explanation for the apparent small synthesis of organic matter in the pyrolytic release experiment remains obscure [76]”.
5. Nontronite and Related Iron-Rich Smectites
5.1. Nontronite and Related Iron-Rich Smectite Synthesis
5.2. Effect of Small Organic Molecules on the Synthesis of Smectites
5.3. Smectites: Catalytic Organic Reactions and Other Organic Interactions
6. Smectite and the Origin of Life
6.1. Simple Biomolecules
6.2. Chirality
6.3. Adsorption on Clay Minerals
6.4. Polymerization of Biomolecules
6.5. Clay Minerals as Potential Information Carriers
6.6. Encapsulation
7. The Clay Hypothesis and the Origin of Life, a Biochemist’s View
7.1. Clay Replication
7.2. Clay Interactions with Peptides
7.3. Clay Minerals on Carbonaceous Chondrites
7.4. Clay Surface Analysis with Atomic Force Microscopy
7.5. Low-Temperature Clay Synthesis and Replication
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dyson, F. Origins of Life, 2nd ed.; Cambridge University Press: Cambridge, UK, 1999; p. 112. [Google Scholar] [CrossRef]
- Miller, S.L. Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef]
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Oparin, A.I. Proiskhozhdenie zhizni; Izd. Moskovskii Rabochii: Moscow, Russia, 1924. [Google Scholar]
- Haldane, J.B.S. Origin of Life. Ration. Ann. 1929, 148, 3–10. [Google Scholar]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The Hypercycle: A Principle of Natural Self-Organization; Springer: Berlin/Heidelberg, Germany, 1979; p. 92. [Google Scholar]
- Orgel, L.E. Evolution of the genetic apparatus. J. Mol. Biol. 1968, 38, 381–393. [Google Scholar] [CrossRef]
- Cairns-Smith, A.G. The origin of life and the nature of the primitive gene. J. Theor. Biol. 1966, 10, 53–88. [Google Scholar] [CrossRef]
- Hartman, H. Speculations on the origin and evolution of metabolism. J. Mol. Evol. 1975, 4, 359–370. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.; Carlson, R.W.; Francis, D.; Stevenson, R.K. Neodynium-142 evidence for Hadean mafic crust. Science 2008, 321, 1828–1831. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.F. On the emergence of plate tectonics. Geology 1992, 20, 963–966. [Google Scholar] [CrossRef]
- Arndt, N.T. Why was flood volcanism on submerged continental platforms so common in the Precambrian? Precambrian Res. 1999, 97, 155–164. [Google Scholar] [CrossRef]
- Macleod, G.; McKeown, C.; Hall, A.J.; Russell, M.J. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig. Life Evol. Biosph. 1994, 24, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Arndt, N.; Lesher, C.M.; Barnes, S.J. Komatiite; Cambridge University Press: Cambridge, UK, 2008; p. 467. [Google Scholar] [CrossRef]
- Arndt, N.T.; Naldrett, A.J.; Pyke, D.R. Komatiitic and Iron-rich tholeiitic lavas of Munro Township, Northeast Ontario. J. Petrol. 1977, 18, 319–369. [Google Scholar] [CrossRef]
- Cockell, C.S. The origin and emergence of life under impact bombardment. Philos. Trans. R. Soc. B 2006, 361, 1845–1856. [Google Scholar] [CrossRef]
- Blake, D.F.; Morris, R.V.; Kocurek, G.; Morrison, S.M.; Downs, R.T.; Bish, D.; Ming, D.W.; Edgett, K.S.; Rubin, D.; Goetz, W.; et al. Curiosity at Gale Crater, Mars: Characterization and Analysis of the Rocknest Sand Shadow. Science 2013, 341. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.K.; Castro, A.J.; Clark, B.C.; III, T.P.; Rose, H., Jr.; Keil, K.; Gooding, J.L. The Viking X ray fluorescence experiment: Sampling strategies and laboratory simulations. J. Geophys. Res. 1977, 82, 4595–4624. [Google Scholar] [CrossRef]
- Toulmin, P., III; Baird, A.K.; Clark, B.C.; Keil, K.; Rose, H.J., Jr.; Christian, R.P.; Evans, P.H.; Kelliher, W.C. Geochemical and mineralogical interpretation of the Viking inorganic chemical results. J. Geophys. Res. (1896–1977) 1977, 82, 4625–4634. [Google Scholar] [CrossRef]
- McSween, H.Y.J., Jr.; Taylor, G.J.; Wyat, M.B. Elemental composition of the Martian crust. Science 2009, 324, 736–739. [Google Scholar] [CrossRef]
- Bibring, J.P.; Langevin, Y.; Gendrin, A.; Gondet, B.; Poulet, F.; Berthé, M.; Soufflot, A.; Arvidson, R.; Mangold, N.; Mustard, J.; et al. Mars surface diversity as revealed by the OMEGA/Mars Express observations. Science 2005, 307, 1576–1581. [Google Scholar] [CrossRef]
- Murchie, S.L.; Seelos, F.P.; Hash, C.D.; Humm, D.C.; Malaret, E.; McGovern, J.A.; Choo, T.H.; Seelos, K.D.; Buczkowski, D.L.; Morgan, M.F.; et al. Compact Reconnaissance Imaging Spectrometer for Mars investigation and data set from the Mars Reconnaissance Orbiter’s primary science phase. J. Geophys. Res. Planets 2009, 114, E00D07. [Google Scholar] [CrossRef]
- Poulet, F.; Bibring, J.-P.; Mustard, J.F.; Gendrin, A.; Mangold, N.; Langevin, Y.; Arvidson, R.E.; Gondet, B.; Gomez, C.; Omega Team. Phyllosilicates on Mars and implications for early Martian climate. Nature 2005, 438, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Mustard, J.F.; Murchie, S.L.; Pelkey, S.M.; Ehlmann, B.L.; Milliken, R.E.; Grant, J.A.; Bibring, J.-P.; Poulet, F.; Bishop, J.L.; Noe Dobrea, E.Z.; et al. Hydrated silicate minerals on mars observed by the mars reconnaissance orbiter CRISM instrument. Nature 2008, 454, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Murchie, S.L.; Mustard, J.F.; Ehlmann, B.L.; Milliken, R.E.; Bishop, J.L.; McKeown, N.K.; Noe Dobrea, E.Z.; Seelos, F.P.; Buczkowski, D.L.; Wiseman, S.M.; et al. A synthesis of Martian aqueous mineralogy after 1 Mars year of observations from the Mars Reconnaissance Orbiter. J. Geophys. Res. Planets 2009, 114. [Google Scholar] [CrossRef]
- Bibring, J.-P.; Langevin, Y.; Mustard, J.F.; Poulet, F.; Arvidson, R.; Gendrin, A.; Gondet, B.; Mangold, N.; Pinet, P.; Forget, F. Global mineralogical and aqueous Mars history derived from OMEGA/Mars Express data. Science 2006, 312, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Weitz, C.M.; Bishop, J.L.; Thollot, P.; Mangold, N.; Roach, L.H. Diverse mineralogies in two troughs of Noctis Labyrinthus, Mars. Geology 2011, 39, 899–902. [Google Scholar] [CrossRef]
- Weitz, C.M.; Bishop, J.L.; Baker, L.L.; Berman, D.C. Fresh exposures of hydrous Fe-bearing amorphous silicates on Mars. Geophys. Res. Lett. 2014, 41, 8744–8751. [Google Scholar] [CrossRef]
- Bishop, J.L.; Noe Dobrea, E.Z.; McKeown, N.K.; Parente, M.; Ehlmann, B.L.; Michalski, J.R.; Milliken, R.E.; Poulet, F.; Swayze, G.A.; Mustard, J.F.; et al. Phyllosilicate diversity and past aqueous activity revealed at Mawrth Vallis, Mars. Science 2008, 321, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.; Loizeau, D.; Mangold, N.; Poulet, F.; Bibring, J.-P. Widespread surface weathering on early Mars: A case for a warmer and wetter climate. Icarus 2015, 248, 373–382. [Google Scholar] [CrossRef]
- McLennan, S.M.; Anderson, R.B.; Bell, J.F.; Bridges, J.C.; Calef, F.; Campbell, J.L.; Clark, B.C.; Clegg, S.; Conrad, P.; Cousin, A.; et al. Elemental Geochemistry of Sedimentary Rocks at Yellowknife Bay, Gale Crater, Mars. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Grotzinger, J.P.; Gupta, S.; Malin, M.C.; Rubin, D.M.; Schieber, J.; Siebach, K.; Sumner, D.Y.; Stack, K.M.; Vasavada, A.R.; Arvidson, R.E.; et al. Deposition, exhumation, and paleoclimate of an ancient lake deposit, Gale crater, Mars. Science 2015, 350. [Google Scholar] [CrossRef] [PubMed]
- Grotzinger, J.P.; Sumner, D.Y.; Kah, L.C.; Stack, K.; Gupta, S.; Edgar, L.; Rubin, D.; Lewis, K.; Schieber, J.; Mangold, N.; et al. A habitable Fluvio-Lacustrine environment at Yellowknife Bay, Gale Crater, Mars. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Bristow, T.F.; Bish, D.L.; Vaniman, D.T.; Morris, R.V.; Blake, D.F.; Grotzinger, J.P.; Rampe, E.B.; Crisp, J.A.; Achilles, C.N.; Ming, D.W.; et al. The origin and implications of clay minerals from Yellowknife Bay, Gale crater, Mars. Am. Mineral. 2015, 100, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.; Poulet, F.; Bibring, J.P.; Mangold, N.; Murchie, S. Hydrous minerals on Mars as seen by the CRISM and OMEGA imaging spectrometers: Updated global view. J. Geophys. Res. Planets 2013, 118, 831–858. [Google Scholar] [CrossRef]
- Ehlmann, B.L.; Mustard, J.F.; Murchie, S.L.; Bibring, J.P.; Meunier, A.; Fraeman, A.A.; Langevin, Y. Subsurface water and clay mineral formation during the early history of Mars. Nature 2011, 479, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.R.; Cuadros, J.; Bishop, J.L.; Darby Dyar, M.; Dekov, V.; Fiore, S. Constraints on the crystal-chemistry of Fe/Mg-rich smectitic clays on Mars and links to global alteration trends. Earth Planet. Sci. Lett. 2015, 427, 215–225. [Google Scholar] [CrossRef]
- Wasson, J.T. Meteorites: Their Record of Early Solar-System History; Freeman: New York, NY, USA, 1985; p. 267. [Google Scholar]
- Lauretta, D.S.; McSween, H.Y., Jr. (Eds.) Meteorites and the Early Solar System II; The University of Arizona Press: Tucson, AZ, USA, 2006; p. 942. [Google Scholar]
- Papike, J.J. (Ed.) Planetary Materials; Mineralogical Society of America: Washington, DC, USA, 1989; Volume 36, p. 1059. [Google Scholar]
- Dominik, B.; Jessberger, E.K.; Staudacher, T.; Nagel, K.; El Goresy, A. A new type of white inclusion in allende—petrography, mineral chemistry, 40Ar–39Ar ages and genetic implications. In Proceedings of the 9th Lunar Planet Science Conference, Houston, TX, USA, 13–17 March 1978; pp. 1249–1266. [Google Scholar]
- Nozette, S.; Wilkening, L.L. Evidence for aqueous alteration in a carbonaceous xenolith from the Plainview (H5) chondrite. Geochim. Cosmochim. Acta 1982, 46, 557–563. [Google Scholar] [CrossRef]
- Cohen, R.E.; Kornacki, A.S.; Wood, J.A. Mineralogy and petrology of chondrules and inclusions in the Mokoia CV3 chondrite. Geochim. Cosmochim. Acta 1983, 47, 1739–1757. [Google Scholar] [CrossRef]
- Morlok, A.; Bischoff, A.; Stephan, T.; Floss, C.; Zinner, E.; Jessberger, E.K. Brecciation and chemical heterogeneities of CI chondrites. Geochim. Cosmochim. Acta 2006, 70, 5371–5394. [Google Scholar] [CrossRef]
- Brown, P.G.; Hildebrand, A.R.; Zolensky, M.E.; Grady, M.; Clayton, R.N.; Mayeda, T.K.; Tagliaferri, E.; Spalding, R.; Macrae, N.D.; Hoffman, E.L.; et al. The fall, recovery, orbit, and composition of the Tagish Lake Meteorite: A new type of carbonaceous chondrite. Science 2000, 290, 320–325. [Google Scholar] [CrossRef]
- McSween, H.Y.J., Jr.; Treiman, A.H. Martian meteorites. In Planet. Mater; Papike, J.J., Ed.; Mineralogical Society of America: Washington, DC, USA, 1998; Volume 36, pp. 6:1–6:53. [Google Scholar]
- Treiman, A.H.; Barrett, R.A.; Gooding, J.L. Preterrestrial aqueous alteration of the Lafayette (SNC) meteorite. Meteoritics 1993, 28, 86–97. [Google Scholar] [CrossRef]
- Bridges, J.C.; Schwenzer, S.P. The nakhlite hydrothermal brine on Mars. Earth Planet. Sci. Lett. 2012, 359–360, 117–123. [Google Scholar] [CrossRef]
- Jones, T.D.; Lebofsky, L.A.; Lewis, J.S.; Marley, M.S. The composition and origin of the C, P, and D asteroids: Water as a tracer of thermal evolution in the outer belt. Icarus 1990, 88, 172–192. [Google Scholar] [CrossRef]
- Tholen, D.J.; Barucci, M.A. Asteroid taxonomy. In Asteroids II; Binzel, R.P., Gehrels, T., Shapley Matthews, M., Eds.; University of Arizona Press: Tucson, AZ, USA, 1989; pp. 298–315. [Google Scholar]
- Bell, J.F.; Davis, D.R.; Hartmann, W.K.; Gaffey, M.J. The big picture. In Asteroids II; Binzel, R.P., Gehrels, T., Shapley Matthews, M., Eds.; University of Arizona Press: Tucson, AZ, USA, 1989; pp. 921–945. [Google Scholar]
- Rivkin, A.S.; Howell, E.S.; Vilas, F.; Lebofsky, L.A. Hydrated Minerals on Asteroids: The Astronomical Record. In Asteroids III; Bottke, W.F., Jr., Cellino, A., Paolicchi, P., Binzel, R.P., Eds.; University of Arizona Press: Tucson, AZ, USA, 2002; pp. 235–253. [Google Scholar]
- McCord, T.; Gaffey, M.J. Asteroids: Surface composition from reflectance spectroscopy. Science 1974, 186, 352–355. [Google Scholar] [CrossRef]
- McCord, T.B.; Sotin, C. Ceres: Evolution and current state. J. Geophys. Res. Planets 2005, 110, E05009. [Google Scholar] [CrossRef]
- Lebofsky, L.A.; Feierberg, M.A.; Tokunaga, A.T.; Larson, H.P.; Johnson, J.R. The 1.7- to 4.2-micron spectrum of asteroid 1 Ceres—evidence for structural water in clay minerals. Icarus 1981, 48, 453–459. [Google Scholar] [CrossRef]
- King, T.V.V.; Clark, R.N.; Calvin, W.M.; Sherman, D.M.; Brown, R.H. Evidence for ammonium-bearing minerals on Ceres. Science 1992, 255, 1551–1553. [Google Scholar] [CrossRef] [PubMed]
- Rivkin, A.S.; Davies, J.K.; Johnson, J.R.; Ellison, S.L.; Trilling, D.E.; Brown, R.H.; Lebofsky, L.A. Hydrogen concentrations on C-class asteroids derived from remote sensing. Meteor. Planet. Sci. 2003, 38, 1383–1398. [Google Scholar] [CrossRef]
- Rivkin, A.S.; Volquardsen, E.L.; Clark, B.E. The surface composition of Ceres: Discovery of carbonates and iron-rich clays. Icarus 2006, 185, 563–567. [Google Scholar] [CrossRef]
- Berg, B.L.; Cloutis, E.A.; Beck, P.; Vernazza, P.; Bishop, J.L.; Driss, T.; Reddy, V.; Applin, D.; Mann, P. Reflectance spectroscopy (0.35–25 mm) of ammonium-bearing minerals and comparison to Ceres family asteroids. Icarus 2016, 265, 218–237. [Google Scholar] [CrossRef]
- Werner, M.W.; Roellig, T.L.; Low, F.J.; Rieke, G.H.; Rieke, M.; Hoffmann, W.F.; Young, E.; Houck, J.R.; Brandl, B.; Fazio, G.G.; et al. The Spitzer Space Telescope mission. Astrophys. J. Suppl. S. 2004, 154, 1–9. [Google Scholar] [CrossRef]
- Lisse, C.M.; Vancleve, J.; Adams, A.C.; A’Hearn, M.F.; Fernández, Y.R.; Farnham, T.L.; Armus, L.; Grillmair, C.J.; Ingalls, J.; Belton, M.J.S.; et al. Spitzer spectral observations of the deep impact ejecta. Science 2006, 313, 635–640. [Google Scholar] [CrossRef]
- Kelley, M.S.; Wooden, D.H. The composition of dust in Jupiter-family comets inferred from infrared spectroscopy. Planet. Space Sci. 2009, 57, 1133–1145. [Google Scholar] [CrossRef]
- Lisse, C.M.; Kraemer, K.E.; Nuth III, J.A.; Li, A.; Joswiak, D. Comparison of the composition of the Tempel 1 ejecta to the dust in Comet C/Hale–Bopp 1995 O1 and YSO HD 100546. Icarus 2007, 187, 69–86. [Google Scholar] [CrossRef]
- Williams, D.M.; Mason, C.G.; Gehrz, R.D.; Jones, T.J.; Charles, E.W.; Harker, D.E.; Hanner, M.S.; Wooden, D.H.; Witteborn, F.C.; Butner, H.M. Measurement of Submicron Grains in the Coma of Comet Hale-Bopp C/1995 O1 during 1997 February 15–20 UT. Astrophys. J. 1997, 489, L91–L94. [Google Scholar] [CrossRef]
- Waelkens, C.; Malfait, K.; Waters, L.B.F.M. Comet Hale-Bopp, circumstellar dust, and the interstellar medium. Earth Moon Planets 1997, 79, 265–274. [Google Scholar] [CrossRef]
- Levin, G.V.; Straat, P.A. The Case for Extant Life on Mars and Its Possible Detection by the Viking Labeled Release Experiment. Astrobiology 2016, 16, 798–810. [Google Scholar] [CrossRef]
- Chambers, P. Life on Mars: The Complete Story; Blandford: London, UK, 1999. [Google Scholar]
- Levin, G.; Straat, P. Viking Labeled Release Biology Experiment: Interim Results. Science 1976, 194, 1322–1329. [Google Scholar] [CrossRef]
- Levin, G.V.; Straat, P.A. Completion of the Viking labeled release experiment on Mars. J. Mol. Evol. 1979, 14, 167–183. [Google Scholar] [CrossRef]
- Miller, J.; Straat, P.; Levin, G. Periodic Analysis of the Viking Lander Labeled Release Experiment; SPIE: Bellingham, WA, USA, 2002; Volume 4495. [Google Scholar]
- Bianciardi, G.; Miller, J.D.; Straat, P.A.; Levin, G.V. Complexity Analysis of the Viking Labeled Release Experiments. Int. J. Aeronaut. Space Sci. 2012, 13, 14–26. [Google Scholar] [CrossRef]
- Horowitz, N.H.; Hobby, G.L.; Hubbard, J.S. The viking carbon assimilation experiments: Interim report. Science 1976, 194, 1321–1322. [Google Scholar] [CrossRef]
- Klein, H.P.; Horowitz, N.H.; Levin, G.V.; Oyama, V.I.; Lederberg, J.; Rich, A.; Hubbard, J.S.; Hobby, G.L.; Straat, P.A.; Berdahl, B.J.; et al. The viking biological investigation: Preliminary results. Science 1976, 194, 99–105. [Google Scholar] [CrossRef]
- Schuerger, A.; Clark, B. Viking Biology Experiments: Lessons Learned and the Role of Ecology in Future Mars Life-Detection Experiments. Space Sci. Rev. 2008, 135, 233–243. [Google Scholar] [CrossRef]
- Klein, H. The Viking biological experiments on Mars. Icarus 1978, 34, 666–674. [Google Scholar] [CrossRef]
- Caplinger, M. Life on Mars. Available online: https://www.msss.com/http/ps/life/life.html (accessed on 8 November 2021).
- Beegle, L.W.; Wilson, M.G.; Abilleira, F.; Jordan, J.F.; Wilson, G.R. A concept for NASA’s Mars 2016 astrobiology field laboratory. Astrobiology 2007, 7, 545–577. [Google Scholar] [CrossRef] [PubMed]
- NASA/Jet Propulsion Laboratory, Martian Life Or Not? NASA’s Phoenix Team Analyzes Results (6 August 2008). Available online: https://www.sciencedaily.com/releases/2008/08/080805192122.htm (accessed on 8 November 2021).
- Navarro-González, R.; Vargas, E.; de la Rosa, J.; Raga, A.C.; McKay, C.P. Reanalysis of the Viking results suggests perchlorate and organics at midlatitudes on Mars. J. Geophys. Res. Planets 2010, 115, E12010, Erratum in J. Geophys. Res. Planets 2011, 116, E08011. [Google Scholar] [CrossRef]
- Biemann, K.; Bada, J.L. Comment on “Reanalysis of the Viking results suggests perchlorate and organics at midlatitudes on Mars” by Rafael Navarro-González et al. J. Geophys. Res. 2011, 116, E12001. [Google Scholar] [CrossRef]
- SPIE. Gilbert Levin: Mars microbes—Proof from the Viking Missions? Available online: https://www.spie.org/news/levin-video?SSO=1 (accessed on 8 November 2021).
- Quinn, R.; Zent, A. Peroxide-Modified Titanium Dioxide: A Chemical Analog of Putative Martian Soil Oxidants. J. Orig. Life Evol. Biosph. 1999, 29, 59–72. [Google Scholar] [CrossRef]
- Levin, G.V. Analysis of evidence of Mars life. Electroneurobiology 2007, 15, 39–44. [Google Scholar]
- Navarro-González, R.; Navarro, K.F.; Rosa, J.d.l.; Iñiguez, E.; Molina, P.; Miranda, L.D.; Morales, P.; Cienfuegos, E.; Coll, P.; Raulin, F.; et al. The limitations on organic detection in Mars-like soils by thermal volatilization–gas chromatography–MS and their implications for the Viking results. Proc. Nat. Acad. Sci. USA 2006, 103, 16089–16094. [Google Scholar] [CrossRef]
- Mars Pathfinder. Available online: https://en.wikipedia.org/wiki/Mars_Pathfinder (accessed on 9 November 2021).
- Spirit (Rover). Available online: https://en.wikipedia.org/wiki/Spirit_(rover) (accessed on 9 November 2021).
- Opportunity (Rover). Available online: https://en.wikipedia.org/wiki/Opportunity_(rover) (accessed on 9 November 2021).
- Phoenix (Spacecraft). Available online: https://en.wikipedia.org/wiki/Phoenix_(spacecraft) (accessed on 9 November 2021).
- Curiosity (Rover). Available online: https://en.wikipedia.org/wiki/Curiosity_(rover) (accessed on 9 November 2021).
- InSight. Available online: https://en.wikipedia.org/wiki/InSight (accessed on 9 November 2021).
- Perseverance (Rover). Available online: https://en.wikipedia.org/wiki/Perseverance_(rover) (accessed on 9 November 2021).
- Tianwen-1. Available online: https://en.wikipedia.org/wiki/Tianwen-1#Scientific_instruments (accessed on 9 November 2021).
- ExoMars. Available online: https://en.wikipedia.org/wiki/ExoMars (accessed on 9 November 2021).
- McKay, C.P.; Stoker, C.R.; Glass, B.J.; Davé, A.I.; Davila, A.F.; Heldmann, J.L.; Marinova, M.M.; Fairen, A.G.; Quinn, R.C.; Zacny, K.A.; et al. The Icebreaker Life Mission to Mars: A Search for Biomolecular Evidence for Life. Astrobiology 2013, 13, 334–353. [Google Scholar] [CrossRef]
- McKay, C.P.; Stoker, C.R.; Glass, B.J.; Davé, A.I.; Davila, A.F.; Heldmann, J.L.; Marinova, M.M.; Fairen, A.G.; Quinn, R.C.; Zacny, K.A.; et al. The Icebreaker Life Mission to Mars: A Search for Biochemical Evidence for Life. In Proceedings of the Concepts and Approaches for Mars Exploration, Lunar and Planetary Institute, Houston, TX, USA, 12–14 June 2012; p. 4091. [Google Scholar]
- Davé, A.; Thompson, S.J.; McKay, C.P.; Stoker, C.R.; Zacny, K.; Paulsen, G.; Mellerowicz, B.; Glass, B.J.; Willson, D.; Bonaccorsi, R.; et al. The Sample Handling System for the Mars Icebreaker Life Mission: From Dirt to Data. Astrobiology 2013, 13, 354–369. [Google Scholar] [CrossRef]
- Parro, V.; Rivas, L.A.; Sebastián, E.; Blanco, Y.; Rodríguez-Manfredi, J.A.; de Diego-Castilla, G.; Moreno-Paz, M.; García-Villadangos, M.; Compostizo, C.; Herrero, P.L.; et al. The Solid3 (“Signs Of Life Detector”) instrument: An antibody microarray-based biosensor for planetary exploration. In Proceedings of the Concepts and Approaches for Mars Exploration, Lunar and Planetary Institute, Houston, TX, USA, 12–14 June 2012; p. 4065. [Google Scholar]
- Hunten, D.M. Possible oxidant sources in the atmosphere and surface of Mars. J. Mol. Evol. 1979, 14, 71–78. [Google Scholar] [CrossRef]
- Bullock, M.A.; Stoker, C.R.; McKay, C.P.; Zent, A.P. A Coupled Soil-Atmosphere Model of H2O2 on Mars. Icarus 1994, 107, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Zolotov, M.Y.; Shock, E.L. Formation of jarosite-bearing deposits through aqueous oxidation of pyrite at Meridiani Planum, Mars. Geophys. Res. Lett. 2005, 32, L21203. [Google Scholar] [CrossRef]
- Fairén, A.G.; Fernández-Remolar, D.; Dohm, J.M.; Baker, V.R.; Amils, R. Inhibition of carbonate synthesis in acidic oceans on early Mars. Nature 2004, 431, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Mottola, H.A.; Simpson, B.E.; Gorin, G. Absorptiometric determination of hydrogen peroxide in submicrogram amounts with leuco crystal violet and peroxidase as catalyst. Anal. Chem. 1970, 42, 410–411. [Google Scholar] [CrossRef]
- Cohn, C.A.; Pak, A.; Strongin, D.; Schoonen, M.A. Quantifying hydrogen peroxide in iron-containing solutions using leuco crystal violet. Geochem. Trans. 2005, 6, 47. [Google Scholar] [CrossRef]
- Schulze-Makuch, D.; Houtkooper, J.; Knoblauch, M.; Furfaro, R.; Fink, W.; Fairén, A.; Vali, H.; Head, J.; Lim, D.S.S.; Dohm, J.; et al. The Biological Oxidant and Life Detection (BOLD) Mission: An Outline for a New Mission to Mars; SPIE: Bellingham, WA, USA, 2007; Volume 6694. [Google Scholar]
- Frost, R.L.; Ding, Z.; Kloprogge, J.T. Does life exist on mars? Do nontronites hold the key to life on mars? Acta Univ. Carol.-Geol. 2002, 46, 28–29. [Google Scholar]
- Frost, R.L.; Kloprogge, J.T.; Ding, Z. Near-infrared spectroscopic study of nontronites and ferruginous smectite. Spectrochim. Acta A 2002, 58, 1657–1668. [Google Scholar] [CrossRef]
- Banin, A.; Rishpon, J. Smectite clays in Mars soil: Evidence for their presence and role in Viking biology experimental results. J. Mol. Evol. 1979, 14, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.L.; Kloprogge, J.T.; Ding, Z. The Garfield and Uley nontronites—An infrared spectroscopic comparison. Spectrochim. Acta A 2002, 58, 1881–1894. [Google Scholar] [CrossRef]
- Catling, D.C.; Moore, J.M. The nature of coarse-grained crystalline hematite and its implications for the early environment of Mars. Icarus 2003, 165, 277–300. [Google Scholar] [CrossRef]
- Catalano, J.G. Thermodynamic and mass balance constraints on iron-bearing phyllosilicate formation and alteration pathways on early Mars. J. Geophys. Res. Planets 2013, 118, 2124–2136. [Google Scholar] [CrossRef]
- Sholes, S.F.; Smith, M.L.; Claire, M.W.; Zahnle, K.J.; Catling, D.C. Anoxic atmospheres on Mars driven by volcanism: Implications for past environments and life. Icarus 2017, 290, 46–62. [Google Scholar] [CrossRef]
- Guven, N. Smectites. In Hydrous Phyllosilicates; Bailey, S.W., Ed.; Mineralogical Society of America: Chelsea, MI, USA, 1988; Volume 19, pp. 497–559. [Google Scholar]
- Burns, R.G. Rates and mechanisms of chemical weathering of ferromagnesian silicate minerals on Mars. Geochim. Cosmochim. Acta 1993, 57, 4555–4574. [Google Scholar] [CrossRef]
- Poulet, F.; Mangold, N.; Loizeau, D.; Bibring, J.-P.; Langevin, Y.; Michalski, J.; Gondet, B. Abundance of minerals in the phyllosilicate-rich units on Mars. Astron. Astrophys. 2008, 487, L41–L44. [Google Scholar] [CrossRef]
- Ehlmann, B.L.; Edwards, C.S. Mineralogy of the martian surface. Ann. Rev. Earth Plan. Sci. 2014, 42, 291–315. [Google Scholar] [CrossRef]
- Arvidson, R.E.; Squyres, S.W.; Bell, J.F., 3rd; Catalano, J.G.; Clark, B.C.; Crumpler, L.S.; de Souza, P.A., Jr.; Fairén, A.G.; Farrand, W.H.; Fox, V.; et al. Ancient aqueous environments at Endeavour crater, Mars. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Fox, V.K.; Arvidson, R.E.; Guinness, E.A.; McLennan, S.M.; Catalano, J.G.; Murchie, S.L.; Powell, K.E. Smectite deposits in Marathon Valley, Endeavour Crater, Mars, identified using CRISM hyperspectral reflectance data. Geophys. Res. Lett. 2016, 43. [Google Scholar] [CrossRef]
- Vaniman, D.T.; Bish, D.L.; Ming, D.W.; Bristow, T.F.; Morris, R.V.; Blake, D.F.; Chipera, S.J.; Morrison, S.M.; Treiman, A.H.; Rampe, E.B.; et al. Mineralogy of a mudstone at Yellowknife Bay, Gale crater, Mars. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Bristow, T.F.; Rampe, E.B.; Achilles, C.N.; Blake, D.F.; Chipera, S.J.; Craig, P.; Crisp, J.A.; Des Marais, D.J.; Downs, R.T.; Gellert, R.; et al. Clay mineral diversity and abundance in sedimentary rocks of Gale crater, Mars. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Rampe, E.B.; Ming, D.W.; Blake, D.F.; Bristow, T.F.; Chipera, S.J.; Grotzinger, J.P.; Morris, R.V.; Morrison, S.M.; Vaniman, D.T.; Yen, A.S.; et al. Mineralogy of an ancient lacustrine mudstone succession from the Murray formation, Gale crater, Mars. Earth Planet. Sci. Lett. 2017, 471, 172–185. [Google Scholar] [CrossRef]
- Sheppard, R.Y.; Thorpe, M.T.; Fraeman, A.A.; Fox, V.K.; Milliken, R.E. Merging Perspectives on Secondary Minerals on Mars: A Review of Ancient Water-Rock Interactions in Gale Crater Inferred from Orbital and In-Situ Observations. Minerals 2021, 11, 986. [Google Scholar] [CrossRef]
- Liu, J.; Michalski, J.R.; Zhou, M.-F. Intense subaerial weathering of eolian sediments in Gale crater, Mars. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Bunch, T.E.; Chang, S. Carbonaceous chondrites—II. Carbonaceous chondrite phyllosilicates and light element geochemistry as indicators of parent body processes and surface conditions. Geochim. Cosmochim. Acta 1980, 44, 1543–1577. [Google Scholar] [CrossRef]
- Ammannito, E.; DeSanctis, M.C.; Ciarniello, M.; Frigeri, A.; Carrozzo, F.G.; Combe, J.-P.; Ehlmann, B.L.; Marchi, S.; McSween, H.Y.; Raponi, A.; et al. Distribution of phyllosilicates on the surface of Ceres. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Fairén, A.G.; Chevrier, V.; Abramov, O.; Marzo, G.A.; Gavin, P.; Davila, A.F.; Tornabene, L.L.; Bishop, J.L.; Roush, T.L.; Gross, C.; et al. Noachian and more recent phyllosilicates in impact craters on Mars. Proc. Nat. Acad. Sci. USA 2010, 107, 12095–12100. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.; Chevrier, V. Thermal alteration of nontronite and montmorillonite: Implications for the martian surface. Icarus 2010, 208, 721–734. [Google Scholar] [CrossRef]
- Frost, R.L.; Ruan, H.; Kloprogge, J.T.; Gates, W.P. Dehydration and dehydroxylation of nontronites and ferruginous smectite. Thermochim. Acta 2000, 346, 63–72. [Google Scholar] [CrossRef]
- Bristow, T.F.; Milliken, R.E. Terrestrial perspective on authigenic clay mineral production in ancient lakes. Clays Clay Miner. 2011, 59, 339–358. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Komarneni, S.; Amonette, J.E. Synthesis of smectite clay minerals: A critical review. Clays Clay Miner. 1999, 47, 529–554. [Google Scholar] [CrossRef]
- Caillère, S.; Henin, S.; Esquevin, J. Synthèses à basse tempèrature de phyllite ferrifère. Compt. Rend Acad. Sci. 1953, 237, 1724–1726. [Google Scholar]
- Caillère, S.; Henin, S.; Esquevin, J. Synthèses à basse tempèrature de quelque minèraux ferrifère (silicates et oxydes). Bull. Soc. Fr. Miner. Cryst. 1955, 78, 227–241. [Google Scholar] [CrossRef]
- Harder, H. Nontronite synthesis at low temperatures. Chem. Geol. 1976, 18, 169–180. [Google Scholar] [CrossRef]
- Harder, H. Synthesis of iron layer silicate minerals under natural conditions. Clays Clay Miner. 1978, 26, 65–72. [Google Scholar] [CrossRef]
- Decarreau, A.; Bonnin, D. Synthesis and crystallogenesis at low temperature of Fe(III)-smectites by evolution of coprecipitated gels: Experiments in partially reducing conditions. Clay Miner. 1986, 21, 861–877. [Google Scholar] [CrossRef]
- Decarreau, A.; Bonnin, D.; Badaut-Trauth, D.; Couty, R.; Kaiser, R. Synthesis and crystallogenesis of ferric smectite by evolution of Si-Fe coprecipitates in oxidizing conditions. Clays Clay Miner. 1987, 22, 207–223. [Google Scholar] [CrossRef]
- Harder, H. The role of magnesium in the formation of smectite minerals. Chem. Geol. 1972, 10, 31–39. [Google Scholar] [CrossRef]
- Harder, H. Clay mineral formation under lateritic weathering conditions. Clay Miner. 1977, 12, 281–288. [Google Scholar] [CrossRef]
- Decarreau, A. Cristallogènese expérimentale des smectites magnésiennes: Hectorite, stévensite. Bull. Mineral. 1980, 103, 579–590. [Google Scholar] [CrossRef]
- Ewell, R.H.; Insley, H. Hydrothermal synthesis of kaolinite, dickite, beidellite and nontronite. J. Res. Nat. Bur. Stand. 1935, 15, 173–186. [Google Scholar] [CrossRef]
- Hamilton, G.; Furtwängler, W. Synthese von Nontronit. Tschermaks Miner. Petrogr. Mitt. 1951, 2, 397–406. [Google Scholar] [CrossRef]
- Mizutani, T.; Fukushima, Y.; Okada, A.; Kamigaito, O.; Kobayashi, T. Synthesis of 1:1 and 2:1 iron phyllosilicates and characterization of their iron state by Mössbauer spectroscopy. Clays Clay Miner. 1991, 39, 381–386. [Google Scholar] [CrossRef]
- Nagase, T.; Iwasaki, T.; Ebina, T.; Hayashi, H.; Onodera, Y.; Chandra Dutta, N. Hydrothermal synthesis of Fe-montmorillonite in Si-Fe-Mg system. Chem. Lett. 1999, 28, 303–304. [Google Scholar] [CrossRef]
- Grauby, O.; Petit, S.; Decarreau, A.; Baronnet, A. The nontronite–saponite series: An experimental approach. Eur. J. Mineral. 1994, 6, 99–112. [Google Scholar] [CrossRef]
- Petit, S.; Decarreau, A.; Gates, W.; Andrieux, P.; Grauby, O. Hydrothermal synthesis of dioctahedral smectites: The Al–Fe3+ chemical series. Part II: Crystal chemistry. Appl. Clay Sci. 2015, 104, 96–105. [Google Scholar] [CrossRef]
- Decarreau, A.; Petit, S.; Martin, F.; Farges, F.; Vieillard, P.; Joussein, E. Hydrothermal synthesis, between 75 and 150°C, of high charge ferric nontronites. Clays Clay Miner. 2008, 56, 322–337. [Google Scholar] [CrossRef]
- Andrieux, P.; Petit, S. Hydrothermal synthesis of dioctahedral smectites: The Al-Fe chemical series. Part I: Influence of experimental conditions. Appl. Clay Sci. 2010, 48, 5–17. [Google Scholar] [CrossRef]
- Baldermann, A.; Dohrmann, R.; Kaufhold, S.; Nickel, C.; Leetofsky-Papst, I.; Dietzel, M. The Fe-Mg-saponite solid solution series: A hydrothermal synthesis study. Clay Miner. 2014, 49, 391–415. [Google Scholar] [CrossRef]
- Baron, F. Le Fer Dans Les Smectites: Une Approche Par Synthèse Minérale; Poitiers University: Poitiers, France, 2016. [Google Scholar]
- Baron, F.; Petit, S.; Tertre, E.; Decarreau, A. Influence of aqueous Si and Fe speciation on tetrahedral Fe(III) substitutions in nontronites: A clay synthesis approach. Clays Clay Miner. 2016, 64, 189–203. [Google Scholar] [CrossRef]
- Fox, V.K.; Kupper, R.J.; Ehlmann, B.L.; Catalano, J.G.; Razzell-Hollis, J.; Abbey, W.J.; Schild, D.J.; Nickerson, R.D.; Peters, J.C.; Katz, S.M.; et al. Synthesis and characterization of Fe(III)-Fe(II)-Mg-Al smectite solid solutions and implications for planetary science. Am. Mineral. 2021, 106, 964–982. [Google Scholar] [CrossRef]
- Chemtob, S.M.; Nickerson, R.D.; Morris, R.V.; Agresti, D.G.; Catalano, J.G. Synthesis and structural characterization of ferrous trioctahedral smectites: Implications for clay mineral genesis and detectability on Mars. J. Geophys. Res. Planets 2015, 120, 1119–1140. [Google Scholar] [CrossRef]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Schumann, D.; Hartman, H.; Eberl, D.D.; Sears, S.K.; Hesse, R.; Vali, H. Formation of Replicating Saponite from a Gel in the Presence of Oxalate: Implications for the Formation of Clay Minerals in Carbonaceous Chondrites and the Origin of Life. Astrobiology 2012, 12, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Ponce, C.P.; Kloprogge, J.T. Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review. Life 2020, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Basu, K.; Hartman, H.; Matocha, C.J.; Sears, S.K.; Vali, H.; Guzman, M.I. Catalyzed Synthesis of Zinc Clays by Prebiotic Central Metabolites. Sci. Rep. 2017, 7, 533. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Synthesis and characterization of saponite clays. Am. Mineral. 2005, 90, 931–944. [Google Scholar] [CrossRef]
- Fialho, D.M.; Roche, T.P.; Hud, N.V. Prebiotic Syntheses of Noncanonical Nucleosides and Nucleotides. Chem. Rev. 2020, 120, 4806–4830. [Google Scholar] [CrossRef]
- Menor-Salván, C. From the Dawn of Organic Chemistry to Astrobiology: Urea as a Foundational Component in the Origin of Nucleobases and Nucleotides. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids; Menor-Salván, C., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 85–142. [Google Scholar]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Homogeneous forced hydrolysis of aluminum through the thermal decomposition of urea. J. Coll. Interf. Sci. 2005, 285, 86–93. [Google Scholar] [CrossRef]
- Besselink, R.; Stawski, T.M.; Freeman, H.M.; Hövelmann, J.; Tobler, D.J.; Benning, L.G. Mechanism of Saponite Crystallization from a Rapidly Formed Amorphous Intermediate. Cryst. Growth Des. 2020, 20, 3365–3373. [Google Scholar] [CrossRef]
- Theng, B.K.G. Clay Mineral Catalysis of Organic Reactions; CRC Press: Boca Raton, FL, USA, 2018; p. 440. [Google Scholar]
- Laszlo, P. Chemical Reactions on Clays. Science 1987, 235, 1473–1477. [Google Scholar] [CrossRef]
- Fripiat, J.J.; Cruz-Cumplido, M.I. Clays as Catalysts for Natural Processes. Ann. Rev. Earth Plan. Sci. 1974, 2, 239–256. [Google Scholar] [CrossRef]
- Mortland, M.M.; Raman, K.V. Surface acidity of smectites in relation to hydration, exchangeable cation, and structure. Clays Clay Miner. 1968, 16, 393–398. [Google Scholar] [CrossRef]
- Adams, J.M.; Ballantine, J.A.; Graham, S.H.; Laub, R.J.; Purnell, J.H.; Reid, P.I.; Shaman, W.Y.M.; Thomas, J.M. Selective chemical conversions using sheet silicate intercalates: Low-temperature addition of water to 1-alkenes. J. Catal. 1979, 58, 238–252. [Google Scholar] [CrossRef]
- Nagendrappa, G. Organic Synthesis using Clay Catalysts. Clays for ‘Green Chemistry’. Resonance 2002, 64–77. [Google Scholar] [CrossRef]
- Adams, J.M.; Clement, D.E.; Graham, S.H. Synthesis of methyl-t-butyl ether from methanol and isobutene using a clay catalyst. Clays Clay Miner. 1982, 30, 129–134. [Google Scholar] [CrossRef]
- Adams, J.M.; Clement, D.E.; Graham, S.H. Reactions of alcohols with alkenes over an aluminum-exchanged montmorillonite. Clays Clay Miner. 1983, 31, 129–136. [Google Scholar] [CrossRef]
- Harun, F.W.; Jihadi, N.I.M.; Ramli, S.; Hassan, N.R.A.; Zubir, N.A.M. Esterification of oleic acid with alcohols over Cu-MMT K10 and Fe-MMT K10 as acid catalysts. AIP Conf. Proc. 2018, 1972, 030025. [Google Scholar] [CrossRef]
- Ballantine, J.A.; Purnell, J.H.; Thomas, J.M. Organic reactions in a clay microenvironment. Clay Miner. 1983, 18, 347–356. [Google Scholar] [CrossRef]
- Fishman, D.; Klug, J.T.; Shani, A. α,β-Unsaturated aldehydes; montmorillonite clay K-10, an effective catalyst for the preparation of unsaturated aldehydes via condensation of acetals with vinyl ethers. Synthesis 1981, 1981, 137–138. [Google Scholar] [CrossRef]
- Kaur, N.; Kishore, D. Montmorillonite: An efficient, heterogeneous and green catalyst for organic synthesis. J. Chem. Pharm. Res. 2012, 4, 991–1015. [Google Scholar]
- Badathala, V. Clay Catalysts in Organic Synthesis. Synlett 2004, 2, 388–389. [Google Scholar] [CrossRef][Green Version]
- Ballantine, J.A.; Davies, M.; Purnell, H.; Rayanakorn, M.; Thomas, J.M.; Williams, K.J. Chemical conversions using sheet silicates: Novel intermolecular dehydrations of alcohols to ethers and polymers. J. Chem. Soc. Chem. Comm. 1981, 427–428. [Google Scholar] [CrossRef]
- Vijayakumar, B.; Nagendrappa, G.; Jai Prakash, B.S. Acid Activated Indian Bentonite, an Efficient Catalyst for Esterification of Carboxylic Acids. Catal. Lett. 2009, 128, 183–189. [Google Scholar] [CrossRef]
- Widi, R.K.; Budhyantoro, A.; Riadi, L. Esterification of palmitic acid over acid catalyst from modified bentonite. Int. J. Appl. Chem. 2010, 6, 11–18. [Google Scholar]
- Rezende, M.J.C.; Pereira, M.S.C.; Santos, G.F.N.; Aroeira, G.O.P.; Albuquerque, T.C., Jr.; Suarez, P.A.Z.; Pinto, A.C. Preparation, characterisation and evaluation of brazilian clay-based catalysts for use in esterification reactions. J. Braz. Chem. Soc. 2012, 23, 1209–1215. [Google Scholar] [CrossRef]
- Rezende, M.; Pinto, A. Esterification of fatty acids using acid-activated Brazilian smectite natural clay as a catalyst. Renew. Energy 2016, 92, 171–177. [Google Scholar] [CrossRef]
- Hamdi, J.; Diehl, B.N.; Kilgore, K.; Lomenzo, S.A.; Trudell, M.L. Halloysite-Catalyzed Esterification of Bio-Mass Derived Acids. ACS Omega 2019, 4, 19437–19441. [Google Scholar] [CrossRef]
- Kalita, C.; Bharadwaj, S.K.; Saikia, P. Solvent-free esterification of substituted benzoic acids with alcohols using modified montmorillonite K10 as solid acid catalyst. Int. J. Sci. Tech. Res. 2020, 9, 658–661. [Google Scholar]
- Harrane, A.; Meghabar, R.; Belbachir, M. Polymerization of ε-caprolactone using a montmorillonite clay as catalyst. Des. Monomers Polym. 2005, 8, 11–24. [Google Scholar] [CrossRef][Green Version]
- Kokel, A.; Schäfer, C.; Török, B. Organic Synthesis Using Environmentally Benign Acid Catalysis. Curr. Org. Synth. 2019, 16, 615–649. [Google Scholar] [CrossRef] [PubMed]
- García, A.; Sanchis, R.; Llopis, F.J.; Vázquez, I.; Pico, M.P.; López, M.L.; Álvarez-Serrano, I.; Solsona, B. Ni Supported on Natural Clays as a Catalyst for the Transformation of Levulinic Acid into γ-Valerolactone without the Addition of Molecular Hydrogen. Energies 2020, 13, 3448. [Google Scholar] [CrossRef]
- Hünig, S.; Benzing, E.; Lücke, E. Synthesen mit Enaminen, I. Acylierung mit Carbonsäurechloriden. Chem. Ber. 1957, 90, 2833–2840. [Google Scholar] [CrossRef]
- Choudary, B.M.; Bhaskar, V.; Kantam, M.L.; Rao, K.K.; Raghavan, K.V. Acylation of Amines with Carboxylic Acids: The Atom Economic Protocol Catalysed by Fe(III)-Montmorillonite. Catal. Lett. 2001, 74, 207–211. [Google Scholar] [CrossRef]
- Kumar, B.S.; Dhakshinamoorthy, A.; Pitchumani, K. K10 montmorillonite clays as environmentally benign catalysts for organic reactions. Catal. Sci. Technol. 2014, 4, 2378–2396. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [PubMed]
- Vinogradoff, V.; Remusat, L.; McLain, H.L.; Aponte, J.C.; Bernard, S.; Danger, G.; Dworkin, J.P.; Elsila, J.E.; Jaber, M. Impact of Phyllosilicates on Amino Acid Formation under Asteroidal Conditions. ACS Earth Space Chem. 2020, 4, 1398–1407. [Google Scholar] [CrossRef]
- Marvi, O. Grinding imidation of anhydrides on smectite clays as recyclable and heterogeneous catalysts under solvent-free conditions. J. Chil. Chem. Soc. 2017, 62, 3501–3504. [Google Scholar] [CrossRef]
- Adams, J.M.; Dyer, S.; Martin, K.; Matear, W.A.; McCabe, R.W. Diels–Alder reactions catalysed by cation-exchanged clay minerals. J. Chem. Soc. Perkin Trans. 1 1994, 761–765. [Google Scholar] [CrossRef]
- Zaccheria, F.; Santoro, F.; Iftitah, E.D.; Ravasio, N. Brønsted and Lewis solid acid catalysts in the valorization of citronellal. Catalysts 2018, 8, 410. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Catalytic activity of synthetic saponite clays: Effects of tetrahedral and octahedral composition. J. Catal. 2005, 231, 443–452. [Google Scholar] [CrossRef]
- Shinde, S.H.; Rode, C.V. Friedel–Crafts Alkylation over Zr-Mont Catalyst for the Production of Diesel Fuel Precursors. ACS Omega 2018, 3, 5491–5501. [Google Scholar] [CrossRef] [PubMed]
- Losfeld, G.; Escande, V.; Vidal de la Blache, P.; l’Huillier, L.; Grison, C. Design and performance of supported Lewis acid catalysts derived from metal contaminated biomass for Friedel-Crafts alkylation and acylation. Catal. Today 2012, 189, 111–116. [Google Scholar] [CrossRef]
- Mortland, M.M.; Halloran, L.J. Polymerization of aromatic molecules on smectite. Soil Sci. Soc. Am. J. 1976, 40, 367–370. [Google Scholar] [CrossRef]
- Pinnavaia, T.J.; Hall, P.L.; Cady, S.S.; Mortland, M.M. Aromatic radical cation formation on the intracrystal surfaces of transition metal layer lattice silicates. J. Phys. Chem. 1974, 78, 994–999. [Google Scholar] [CrossRef]
- Isaacson, P.J.; Sawhney, B.L. Sorption and transformation of phenols on clay surfaces: Effect of exchangeable cations. Clay Miner. 1983, 18, 253–265. [Google Scholar] [CrossRef]
- Solomon, D.H.; Loft, B.C.; Swift, J.D. Reactions catalyzed by minerals IV. The mechanism of the benzidine blue reaction on silicate minerals. Clay Miner. 1968, 7, 389–397. [Google Scholar] [CrossRef]
- Wang, M.C.; Huang, P.M. Catalytic polymerization of hydroquinone by nontronite. Can. J. Soil Sci. 1987, 67, 867–875. [Google Scholar] [CrossRef]
- Wang, M.C.; Huang, P.M. Pyrogallol Transformations as Catalyzed by Nontronite, Bentonite, and Kaolinite. Clays Clay Miner. 1989, 37, 525–531. [Google Scholar] [CrossRef]
- Wang, M.C. Catalysis of nontronite in phenols and glycine transformations. Clays Clay Miner. 1991, 39, 202–210. [Google Scholar]
- Cervini-Silva, J.; Wu, J.; Stucki, J.W.; Larson, R.A. Adsorption kinetics of pentachloroethane by iron-bearing smectites. Clays Clay Miner. 2000, 48, 132–138. [Google Scholar] [CrossRef]
- Stucki, J.W.; Tessier, D. Effects of iron oxidation state on the texture and structural order of Na-nontronite gels. Clays Clay Miner. 1991, 39, 137–143. [Google Scholar] [CrossRef]
- Yan, L.; Stucki, J.W. Effects of structural Fe oxidation state on the coupling of interlayer water and structural Si-O stretching vibrations in montmorillonite. Langmuir 1999, 15, 4648–4657. [Google Scholar] [CrossRef]
- Liu, R.; Xiao, D.; Guo, Y.; Wang, Z.; Liu, J. A novel photosensitized Fenton reaction catalyzed by sandwiched iron in synthetic nontronite. RSC Adv. 2014, 4, 12958–12963. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, B.; Wang, W.; Zhang, W.; Du, P.; Liu, W.; Xing, X.; Ding, D.; Lv, G.; Lv, Q.; et al. In situ produced hydrogen peroxide by biosynthesized Palladium nanoparticles and natural clay mineral for Highly-efficient Carbamazepine degradation. Chem. Eng. J. 2021, 426, 131567. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, Y.; Wen, T.; Zhang, T.; Song, S. Synthetic Fe-rich nontronite as a novel activator of bisulfite for the efficient removal of tetracycline. J. Environ. Man. 2022, 302, 114002. [Google Scholar] [CrossRef] [PubMed]
- Gerakines, P.A.; Hudson, R.L. Glycine’s radiolytic destruction in ices: First in situ laboratory measurements for Mars. Astrobiology 2013, 13, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Hintze, P.E.; Buhler, C.R.; Schuerger, A.C.; Calle, L.M.; Calle, C.I. Alteration of five organic compounds by glow discharge plasma and UV light under simulated Mars conditions. Icarus 2010, 208, 749–757. [Google Scholar] [CrossRef]
- Johnson, A.P.; Pratt, L.M. Metal-catalyzed degradation and racemization of amino acids in iron sulfate brines under simulated martian surface conditions. Icarus 2010, 207, 124–132. [Google Scholar] [CrossRef]
- Stoker, C.R.; Bullock, M.A. Organic degradation under simulated Martian conditions. J. Geophys. Res. 1997, 102, 10881–10888. [Google Scholar] [CrossRef]
- Poch, O.; Noblet, A.; Stalport, F.; Correia, J.J.; Grand, N.; Szopa, C.; Coll, P. Chemical evolution of organic molecules under Mars-like UV radiation conditions simulated in the laboratory with the “Mars organic molecule irradiation and evolution” (MOMIE) setup. Planet. Space Sci. 2013, 85, 188–197. [Google Scholar] [CrossRef]
- Poch, O.; Kaci, S.; Stalport, F.; Szopa, C.; Coll, P. Laboratory insights into the chemical and kinetic evolution of several organic molecules under simulated Mars surface UV radiation conditions. Icarus 2014, 242, 50–63. [Google Scholar] [CrossRef]
- Stalport, F.; Coll, P.; Szopa, C.; Raulin, F. Investigating the photostability of carboxylic acids exposed to Mars surface radiation conditions. Astrobiol. 2009, 9, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Ten Kate, I.L.; Garry, J.R.C.; Peeters, Z.; Foing, B.; Ehrenfreund, P. The effects of Martian near surface conditions on the photochemistry of amino acids. Planet. Space Sci. 2006, 54, 296–302. [Google Scholar] [CrossRef]
- Garry, J.R.C.; Ten Kate, I.L.; Martins, Z.; Nornberg, P.; Ehrenfreund, P. Analysis and survival of amino acids in Martian regolith analogs. Meteorit. Planet. Sci. 2006, 41, 391–405. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Chemerisov, S.D.; Marin, T.W. Photocatalytic decomposition of carboxylated molecules on light-exposed Martian regolith and its relation to methane production on Mars. Astrobiol. 2010, 10, 425–436. [Google Scholar] [CrossRef]
- Stalport, F.; Guan, Y.Y.; Coll, P.; Szopa, C.; Macari, F.; Raulin, F.; Chaput, D.; Cottin, H. UVolution, a photochemistry experiment in low Earth orbit: Investigation of the photostability of carboxylic acids exposed to Mars surface UV radiation conditions. Astrobiol. 2010, 10, 449–461. [Google Scholar] [CrossRef]
- Poch, O.; Jaber, M.; Stalport, F.; Nowak, S.; Georgelin, T.; Lambert, J.-F.; Szopa, C.; Coll, P. Effect of nontronite smectite clay on the chemical evolution of several organic molecules under simulated Martian surface ultraviolet radiation conditions. Astrobiology 2015, 15, 221–237. [Google Scholar] [CrossRef]
- Gil-Lozano, C.; Fairén, A.G.; Muñoz-Iglesias, V.; Fernández-Sampedro, M.; Prieto-Ballesteros, O.; Gago-Duport, L.; Losa-Adams, E.; Carrizo, D.; Bishop, J.L.; Fornaro, T.; et al. Constraining the preservation of organic compounds in Mars analog nontronites after exposure to acid and alkaline fluids. Sci. Rep. 2020, 10, 15097. [Google Scholar] [CrossRef]
- Eigenbrode, J.L.; Summons, R.E.; Steele, A.; Freissinet, C.; Millan, M.; Mahaffy, P.R.; Sutter, B.; McAdam, A.; Franz, H.; Archer, P.D. Organic matter in 3.5-billion-year-old mudstones from an ancient lake in Gale Crater, Mars. In Proceedings of the Abstracts of Papers, 257th ACS National Meeting & Exposition, Orlando, FL, USA, 31 March–4 April 2019; p. GEOC-0072. [Google Scholar]
- Freissinet, C.; Glavin, D.P.; Mahaffy, P.R.; Miller, K.E.; Eigenbrode, J.L.; Summons, R.E.; Brunner, A.E.; Buch, A.; Szopa, C.; Archer, P.D., Jr.; et al. Organic molecules in the Sheepbed Mudstone, Gale Crater, Mars. J. Geophys. Res. Planets 2015, 120, 495–514. [Google Scholar] [CrossRef]
- Guzman, M.; McKay, C.P.; Quinn, R.C.; Szopa, C.; Davila, A.F.; Navarro-Gonzalez, R.; Freissinet, C. Identification of Chlorobenzene in the Viking Gas Chromatograph-Mass Spectrometer Data Sets: Reanalysis of Viking Mission Data Consistent With Aromatic Organic Compounds on Mars. J. Geophys. Res. Planets 2018, 123, 1674–1683. [Google Scholar] [CrossRef]
- Hand, E. Mars rover finds long-chain organic compounds. Science 2015, 347, 1402–1403. [Google Scholar] [CrossRef] [PubMed]
- Heinz, J.; Schulze-Makuch, D. Thiophenes on Mars: Biotic or Abiotic Origin? Astrobiology 2020, 20, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Mahaffy, P.R. Discovery of organics on Mars with the SAM experiment on the Curiosity rover. In Proceedings of the Abstracts of Papers, 257th ACS National Meeting & Exposition, Orlando, FL, USA, 31 March–4 April 2019; p. ANYL-0017. [Google Scholar]
- Miller, K.E.; Eigenbrode, J.L.; Freissinet, C.; Glavin, D.P.; Kotrc, B.; Francois, P.; Summons, R.E. Potential precursor compounds for chlorohydrocarbons detected in Gale Crater, Mars, by the SAM instrument suite on the Curiosity Rover. J. Geophys. Res. Planets 2016, 121, 296–308. [Google Scholar] [CrossRef]
- Sutter, B.; McAdam, A.C.; Mahaffy, P.R.; Ming, D.W.; Edgett, K.S.; Rampe, E.B.; Eigenbrode, J.L.; Franz, H.B.; Freissinet, C.; Grotzinger, J.P.; et al. Evolved gas analyses of sedimentary rocks and eolian sediment in Gale Crater, Mars: Results of the Curiosity rover’s sample analysis at Mars instrument from Yellowknife Bay to the Namib Dune. J. Geophys. Res. Planets 2017, 122, 2574–2609. [Google Scholar] [CrossRef]
- Szopa, C.; Freissinet, C.; Glavin, D.P.; Millan, M.; Buch, A.; Franz, H.B.; Summons, R.E.; Sumner, D.Y.; Sutter, B.; Eigenbrode, J.L.; et al. First Detections of Dichlorobenzene Isomers and Trichloromethylpropane from Organic Matter Indigenous to Mars Mudstone in Gale Crater, Mars: Results from the Sample Analysis at Mars Instrument Onboard the Curiosity Rover. Astrobiology 2020, 20, 292–306. [Google Scholar] [CrossRef]
- Voosen, P. NASA Curiosity rover hits organic pay dirt on Mars. Science 2018, 360, 1054–1055. [Google Scholar] [CrossRef]
- Brinton, K.L.F.; Bada, J.L. A reexamination of amino acids in lunar soils: Implications for the survival of exogenous organic material during impact delivery. Geochim. Cosmochim. Acta 1996, 60, 349–354. [Google Scholar] [CrossRef]
- Oro, J.; Updegrove, W.S.; Gibert, J.; McReynolds, J.; Gil-Av, E.; Ibanez, J.; Zlatkis, A.; Flory, D.A.; Levy, R.L.; Wolf, C.J. Organogenic elements and compounds in type C and D lunar samples from Apollo 11. Proc. Apollo 11 [Eleven] Lunar Sci. Conf. 1970, 2, 1901–1920. [Google Scholar]
- Bergantini, A.; Kaiser, R.I. In Situ Detection of Organics in the Comet 67P/Churyumov-Gerasimenko. Chem. Commun. 2016, 1, 824–826. [Google Scholar] [CrossRef][Green Version]
- Elsila, J.E.; Gillette, J.S.; Zare, R.N.; Bernstein, M.P.; Dworkin, J.P.; Sandford, S.A.; Allamandola, L.J. Low temperature ice photochemistry as a source of meteoritic and cometary organics. In Proceedings of the Abstracts of Papers, 221st ACS National Meeting, San Diego, CA, USA, 1–5 April 2001; p. GEOC-151. [Google Scholar]
- Fray, N.; Bardyn, A.; Cottin, H.; Altwegg, K.; Baklouti, D.; Briois, C.; Colangeli, L.; Engrand, C.; Fischer, H.; Glasmachers, A.; et al. High-molecular-weight organic matter in the particles of comet 67P/Churyumov-Gerasimenko. Nature 2016, 538, 72–74. [Google Scholar] [CrossRef]
- Messenger, S.; Nguyen, A.N. Properties and origins of cometary and asteroidal organic matter delivered to the early Earth. In Proceedings of the Abstracts of Papers, 254th ACS National Meeting & Exposition, Washington, DC, USA, 20–24 August 2017; p. PHYS-258. [Google Scholar]
- Schurig, V. Enantiomers on Mars, moons and comets. Nachr. Chem. 2015, 63, 660–664. [Google Scholar] [CrossRef]
- Becker, R.H.; Epstein, S. Carbon, hydrogen and nitrogen isotopes in solvent-extractable organic matter from carbonaceous chondrites. Geochim. Cosmochim. Acta 1982, 46, 97–103. [Google Scholar] [CrossRef]
- Botta, O. Extraterrestrial organic chemistry as recorded in carbonaceous chondrites. ACS Symp. Ser. 2007, 981, 246–260. [Google Scholar]
- Botta, O.; Glavin, D.P.; Bada, J.L. Amino acid signatures in carbonaceous meteorites. Proc. SPIE-Int. Soc. Opt. Eng. 2002, 4495, 27–39. [Google Scholar]
- Burton, A.S.; Elsila, J.E.; Callahan, M.P.; Martin, M.G.; Glavin, D.P.; Johnson, N.M.; Dworkin, J.P. A propensity for n-ω-amino acids in thermally altered Antarctic meteorites. Meteorit. Planet. Sci. 2012, 47, 374–386. [Google Scholar] [CrossRef]
- Changela, H.G.; Le Guillou, C.; Bernard, S.; Brearley, A.J. Hydrothermal evolution of the morphology, molecular composition, and distribution of organic matter in CR (Renazzo-type) chondrites. Meteorit. Planet. Sci. 2018, 53, 1006–1029. [Google Scholar] [CrossRef]
- Cooper, G.; Kimmich, N.; Belisle, W.; Sarlnana, J.; Brabham, K.; Garrel, L. Carbonaceous meteorites as a source of sugar-related organic compounds for the early Earth. Nature 2001, 414, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Derenne, S.; Robert, F. Model of molecular structure of the insoluble organic matter isolated from Murchison meteorite. Meteorit. Planet. Sci. 2010, 45, 1461–1475. [Google Scholar] [CrossRef]
- Elsila, J.E.; Johnson, N.M.; Glavin, D.P.; Aponte, J.C.; Dworkin, J.P. Amino acid abundances and compositions in iron and stony-iron meteorites. Meteorit. Planet. Sci. 2021, 56, 586–600. [Google Scholar] [CrossRef]
- Furukawa, Y.; Chikaraishi, Y.; Ohkouchi, N.; Ogawa, N.O.; Glavin, D.P.; Dworkin, J.P.; Abe, C.; Nakamura, T. Extraterrestrial ribose and other sugars in primitive meteorites. Proc. Natl. Acad. Sci. USA 2019, 116, 24440–24445. [Google Scholar] [CrossRef]
- Glavin, D.P.; Dworkin, J.P. Enrichment of the amino acid L-isovaline by aqueous alteration on CI and CM meteorite parent bodies. Proc. Natl. Acad. Sci. USA 2009, 106, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Hartman, H.; Sweeney, M.A.; Kropp, M.A.; Lewis, J.S. Carbonaceous chondrites and the origin of life. Orig. Life Evol. Biosph. 1993, 23, 221–227. [Google Scholar] [CrossRef]
- Hilts, R.W.; Herd, C.D.K.; Simkus, D.N.; Slater, G.F. Soluble organic compounds in the Tagish Lake meteorite. Meteorit. Planet. Sci. 2014, 49, 526–549. [Google Scholar] [CrossRef]
- Kminek, G.; Botta, O.; Glavin, D.P.; Bada, J.L. Amino acids in the Tagish Lake meteorite. Meteorit. Planet. Sci. 2002, 37, 697–701. [Google Scholar] [CrossRef]
- Martins, Z. Organic chemistry of carbonaceous meteorites. Elements 2011, 7, 35–40. [Google Scholar] [CrossRef]
- Martins, Z. Extraterrestrial organic molecules in meteorites clues to the early solar system. Biochemist 2014, 36, 13–15. [Google Scholar] [CrossRef]
- Monroe, A.A.; Pizzarello, S. The soluble organic compounds of the Bells meteorite: Not a unique or unusual composition. Geochim. Cosmochim. Acta 2011, 75, 7585–7595. [Google Scholar] [CrossRef]
- Moreno-Paz, M.; Gomez-Cifuentes, A.; Ruiz-Bermejo, M.; Hofstetter, O.; Maquieira, A.; Manchado, J.M.; Morais, S.; Sephton, M.A.; Niessner, R.; Knopp, D.; et al. Detecting Nonvolatile Life- and Nonlife-Derived Organics in a Carbonaceous Chondrite Analogue with a New Multiplex Immunoassay and Its Relevance for Planetary Exploration. Astrobiol. 2018, 18, 1041–1056. [Google Scholar] [CrossRef]
- Naraoka, H.; Hashiguchi, M. Distinct distribution of soluble N-heterocyclic compounds between CM and CR chondrites. Geochem. J. 2019, 53, 33–40. [Google Scholar] [CrossRef]
- Naraoka, H.; Yamashita, Y.; Yamaguchi, M.; Orthous-Daunay, F.-R. Molecular Evolution of N-Containing Cyclic Compounds in the Parent Body of the Murchison Meteorite. ACS Earth Space Chem. 2017, 1, 540–550. [Google Scholar] [CrossRef]
- Oba, Y.; Takano, Y.; Naraoka, H.; Furukawa, Y.; Glavin, D.P.; Dworkin, J.P.; Tachibana, S. Extraterrestrial hexamethylenetetramine in meteorites-a precursor of prebiotic chemistry in the inner solar system. Nat. Commun. 2020, 11, 6243. [Google Scholar] [CrossRef] [PubMed]
- Pearson, V.K.; Wilson, R.C.; Gilmour, I. Extraterrestrial organic matter as recorded in meteorites. In Astrobiology, Emergence, Search and Detection of Life; Vladimir, B., Ed.; American Scientific Publishers: Valencia, CA, USA, 2010; pp. 155–173. [Google Scholar]
- Pizzarello, S.; Schrader, D.L.; Monroe, A.A.; Lauretta, D.S. Large enantiomeric excesses in primitive meteorites and the diverse effects of water in cosmochemical evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 11949–11954. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Shock, E.; Shock, E. Carbonaceous Chondrite Meteorites: The Chronicle of a Potential Evolutionary Path between Stars and Life. Orig. Life Evol. Biosph. 2017, 47, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Yarnes, C.T. Enantiomeric excesses of chiral amines in ammonia-rich carbonaceous meteorites. Earth Planet. Sci. Lett. 2016, 443, 176–184. [Google Scholar] [CrossRef]
- Remusat, L.; Le Guillou, C.; Rouzaud, J.-N.; Binet, L.; Derenne, S.; Robert, F. Molecular study of insoluble organic matter in Kainsaz CO3 carbonaceous chondrite: Comparison with CI and CM IOM. Meteorit. Planet. Sci. 2008, 43, 1099–1111. [Google Scholar] [CrossRef]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefuegi, I.; Eckel, G.; Hertkorn, N. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc. Natl. Acad. Sci. USA 2010, 107, 2763–2768. [Google Scholar] [CrossRef]
- Sephton, M.A. Organic compounds in carbonaceous meteorites. Nat. Prod. Rep. 2002, 19, 292–311. [Google Scholar] [CrossRef]
- Sephton, M.A. Organic matter in ancient meteorites. Astron. Geophys. 2004, 45, 8–14. [Google Scholar] [CrossRef]
- Sephton, M.A.; Verchovsky, A.B.; Wright, I.P. Carbon and nitrogen isotope ratios in meteoritic organic matter: Indicators of alteration processes on the parent asteroid. Int. J. Astrobiol. 2004, 3, 221–227. [Google Scholar] [CrossRef][Green Version]
- Sephton, M.A.; Wright, I.P.; Gilmour, I.; de Leeuw, J.W.; Grady, M.M.; Pillinger, C.T. High molecular weight organic matter in martian meteorites. Planet. Space Sci. 2002, 50, 711–716. [Google Scholar] [CrossRef]
- Simkus, D.N.; Aponte, J.C.; Elsila, J.E.; Hilts, R.W.; McLain, H.L.; Herd, C.D.K. New insights into the heterogeneity of the Tagish Lake meteorite: Soluble organic compositions of variously altered specimens. Meteorit. Planet. Sci. 2019, 54, 1283–1302. [Google Scholar] [CrossRef]
- Vinogradoff, V.; Le Guillou, C.; Bernard, S.; Binet, L.; Cartigny, P.; Brearley, A.J.; Remusat, L. Paris vs. Murchison: Impact of hydrothermal alteration on organic matter in CM chondrites. Geochim. Cosmochim. Acta 2017, 212, 234–252. [Google Scholar] [CrossRef]
- Campins, H.; Hargrove, K.; Pinilla-Alonso, N.; Howell, E.S.; Kelley, M.S.; Licandro, J.; Mothe-Diniz, T.; Fernandez, Y.; Ziffer, J. Water ice and organics on the surface of the asteroid 24 Themis. Nature 2010, 464, 1320–1321. [Google Scholar] [CrossRef] [PubMed]
- Duan, A.; Wu, Y.; Cloutis, E.A.; Yu, J.; Li, S.; Jiang, Y. Heating of carbonaceous materials: Insights into the effects of thermal metamorphism on spectral properties of carbonaceous chondrites and asteroids. Meteorit. Planet. Sci. 2021, 56, 2035–2046. [Google Scholar] [CrossRef]
- Pizzarello, S.; Williams, L.B.; Lehman, J.; Holland, G.P.; Yarger, J.L. Abundant ammonia in primitive asteroids and the case for a possible exobiology. Proc. Natl. Acad. Sci. USA 2011, 108, 4303–4306. [Google Scholar] [CrossRef]
- Sabbah, H.; Morrow, A.L.; Jenniskens, P.; Shaddad, M.H.; Zare, R.N. Polycyclic aromatic hydrocarbons in asteroid 2008 TC3: Dispersion of organic compounds inside asteroids. Meteorit. Planet. Sci. 2010, 45, 1710–1717. [Google Scholar] [CrossRef]
- Simakov, M.B. Asteroids and the origin of life-two steps of chemical evolution on the surface of these objects. Earth Planets Space 2008, 60, 75–82. [Google Scholar] [CrossRef]
- Bishop, J.L.; Pieters, C.M.; Hiroi, T.; Mustard, J.F. Spectroscopic analysis of Martian meteorite Allan Hills 84001 powder and applications for spectral identification of minerals and other soil components on Mars. Meteorit. Planet. Sci. 1998, 33, 699–707. [Google Scholar] [CrossRef]
- Jaramillo, E.A.; Royle, S.H.; Claire, M.W.; Kounaves, S.P.; Sephton, M.A. Indigenous Organic-Oxidized Fluid Interactions in the Tissint Mars Meteorite. Geophys. Res. Lett. 2019, 46, 3090–3098. [Google Scholar] [CrossRef]
- Becker, L.; Popp, B.; Rust, T.; Bada, J.L. The origin of organic matter in the Martian meteorite ALH84001. Adv. Space Res. 1999, 24, 477–488. [Google Scholar] [CrossRef]
- Jull, A.J.T.; Courtney, C.; Jeffrey, D.A.; Beck, J.W. Isotopic evidence for a terrestrial source of organic compounds found in Martian meteorites Allan Hills 84001 and Elephant Moraine 79001. Science 1998, 279, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.; McCubbin, F.M.; Fries, M.D. The provenance, formation, and implications of reduced carbon phases in Martian meteorites. Meteorit. Planet. Sci. 2016, 51, 2203–2225. [Google Scholar] [CrossRef]
- Fox, A.C.; Eigenbrode, J.L.; Freeman, K.H. Radiolysis of Macromolecular Organic Material in Mars-Relevant Mineral Matrices. J. Geophys. Res. Planets 2019, 124, 3257–3266. [Google Scholar] [CrossRef]
- Lambert, J.-F. Origins of life: From the mineral to the biochemical world. In Proceedings of the ORIGINS–Studies in Biological and Cultural Evolution, Paris, France, 14 October 2014; p. 00012. [Google Scholar]
- Bernal, J.D. The physical basis of life. Proc. Phys. Soc. A 1949, 62, 537–558. [Google Scholar] [CrossRef]
- Cairns-Smith, A.G. Genetic Takeover: And the Mineral Origins of Life; Cambridge University Press: Cambridge, UK, 1982; p. 477. [Google Scholar]
- Ponnamperuma, C.; Shimoyama, A.; Friebele, E. Clay and the origin of life. Orig. Life 1982, 12, 9–40. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A. Replication and evolution in inorganic systems. Angew. Chem. Int. Ed. 1981, 20, 850–860. [Google Scholar] [CrossRef]
- Arrhenius, G.; Cairns-Smith, A.G.; Hartman, H.; Miller, S.L.; Orgel, L.E. Remarks on the Review Article “Replication and Evolution in Inorganic Systems” by Armin Weiss. Angew. Chem. Int. Ed. 1986, 98, 654. [Google Scholar] [CrossRef]
- Yoshino, D.; Hayatsu, K.; Anders, E. Origin of organic matter in early solar system—III. Amino acids: Catalytic synthesis. Geochim. Cosmochim. Acta 1971, 35, 927–938. [Google Scholar] [CrossRef]
- Shimoyama, A.; Blair, N.; Ponnamperuma, C. Synthesis of amino acids under primitive Earth conditions in the presence of clay. In Origin of Life; Noda, H., Ed.; Center for Academic Publications Japan/Japan Scientific Societies Press: Tokyo, Japan, 1978; pp. 95–99. [Google Scholar]
- Hubbard, J.S.; Hardy, J.P.; Voecks, G.E.; Golub, E.E. Photocatalytic synthesis of organic compounds from CO and water: Involvement of surfaces in the formation and stabilization of products. J. Mol. Evol. 1973, 2, 149–166. [Google Scholar] [CrossRef]
- Cruz, M.; Kaiser, A.; Rouxhet, P.G.; Fripiat, J.J. Adsorption and Transformation of HCN on the Surface of Copper and Calcium Montmorillonite. Clays Clay Miner. 1974, 22, 417–425. [Google Scholar] [CrossRef]
- Ferris, J.P.; Edelson, E.H.; Mount, N.M.; Sullivan, A.E. The effect of clays on the oligomerization of HCN. J. Mol. Evol. 1979, 13, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Degens, E.T.; Matheja, J. Formation of organic polymers on inorganic templates. In Prebiotic Biochemistry Evolution; Kimball, A.P., Oro, J., Eds.; North-Holland Pub. Co.: Amsterdam, The Netherlands, 1971; pp. 39–69. [Google Scholar]
- Chittenden, G.J.F.; Schwartz, A.W. Possible Pathway of Prebiotic Uracil Synthesis by Photodehydrogenation. Nature 1976, 263, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Urey, H.C. Organic Compound Synthesis on the Primitive Earth. Science 1959, 130, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S. Polymerization of Hydrogen Cyanide and Production of Amino Acids and Nucleic Acid Bases in the Presence of Clay Minerals-In Relation to Clay and the Origin of Life. Nendo Kagaku 1989, 29, 89–96. (In Japanese) [Google Scholar]
- Kobayashi, K.; Tsuchiya, M.; Oshima, T.; Yanagawa, H. Abiotic Synthesis of Amino Acids and Imidazole by Proton Irradiation of Similated Primitive Earth Atmospheres. Orig. Life 1990, 20, 99–109. [Google Scholar] [CrossRef]
- Lahav, N.; White, D.; Chang, S. Peptide formation in the prebiotic era: Thermal condensation of glycine in fluctuating clay environments. Science 1978, 201, 67–69. [Google Scholar] [CrossRef]
- Lahav, N.; White, D.H. A possible role of fluctuating clay-water systems in the production of ordered prebiotic oligomers. J. Mol. Evol. 1980, 16, 11–21. [Google Scholar] [CrossRef]
- Bujdák, J.; Slosiariková, H.; Texler, N.; Schwendiger, M.; Rode, B.M. On the possible role of montmorillonite in prebiotic peptide formation. Monatsh. Chem. 1994, 125, 1033–1039. [Google Scholar] [CrossRef]
- Williams, L.B.; Canfield, B.; Voglesonger, K.M.; Holloway, J.R. Organic molecules formed in a “primordial womb”. Geology 2005, 33, 913–916. [Google Scholar] [CrossRef]
- Hashizume, H. Role of Clay Minerals in Chemical Evolution and the Origins of Life. In Clay Minerals in Nature—Their Characterization, Modification and Application; Valaškova, M., Martynkova, G.S., Eds.; IntechOpen: London, UK, 2012. [Google Scholar]
- Friebele, E.; Shimoyama, A.; Hare, P.E.; Ponnamperuma, C. Adsorption of Amino Acid Entantiomers by Na-Montmorillonite. Orig. Life 1981, 11, 173–184. [Google Scholar] [CrossRef]
- Siffert, B.; Naidja, A. Stereoselectivity of montmorillonite in the adsorption and deamination of some amino acids. Clay Miner. 1992, 27, 109–118. [Google Scholar] [CrossRef]
- Hashizume, H.; Theng, B.K.G.; Yamagishi, A. Adsorption and Discrimination of Alanine and Alanyl-Alanine Enantiomers by Allophane. Clay Miner. 2002, 37, 551–557. [Google Scholar] [CrossRef]
- Damien, C.M.; Luthey-Schulten, Z. Influence of montmorillonite on nucleotide oligomerization reactions: A molecular dynamics study. Orig. Life Evol. Biosph. 2010, 40, 303–317. [Google Scholar]
- Bernal, J.D. The Physical Basis of Life; Routledge and Kegan Paul: London, UK, 1951; p. 34. [Google Scholar]
- Henrichs, S.M.; Sugai, S.F. Adsorption of amino acids and glucose by sediments of Resurrection Bay, Alaska, USA: Functional group effects. Geochim. Cosmochim. Acta 1993, 57, 823–835. [Google Scholar] [CrossRef]
- Lahav, N.; Chang, S. The possible role of solid surface area in condensation reactions during chemical evolution: Reevaluation. J. Mol. Evol. 1976, 8, 357–380. [Google Scholar] [CrossRef]
- Fu, L.; Weckhuysen, B.M.; Verberckmoes, A.A.; Schoonheydt, R.A. Clay intercalated Cu(II) amino acid complexes: Synthesis, spectroscopy and catalysis. Clay Miner. 1996, 31, 491–500. [Google Scholar] [CrossRef][Green Version]
- Gupta, A.; Loew, G.H.; Lawless, J. Interaction of metal ions and amino acids: Possible mechanisms for the adsorption of amino acids on homoionic smectite clays. Inorg. Chem. 1983, 22, 111–120. [Google Scholar] [CrossRef]
- Odom, D.G.; Rao, M.; Lawless, J.G.; Oro, J. Association of nucleotides with homoionic clays. J. Mol. Evol. 1979, 12, 365–367. [Google Scholar] [CrossRef]
- Rishpon, J.; O’Hara, P.J.; Lahav, N.; Lawless, J.G. Interaction between ATP, metal ions, glycine, and several minerals. J. Mol. Evol. 1982, 18, 179–184. [Google Scholar] [CrossRef]
- Prabahar, K.J.; Ferris, J.P. Adenine derivatives as phosphate-activating groups for the regioselective formation of 3′,5′-linked oligoadenylates on montmorillonite: Possible phosphate-activating groups for the prebiotic synthesis of RNA. J. Am. Chem. Soc. 1997, 119, 4330–4337. [Google Scholar] [CrossRef]
- Sowerby, S.J.; Cohn, C.A.; Heckl, W.M.; Holm, N.G. Differential adsorption of nucleic acid bases: Relevance to the origin of life. Proc. Nat. Acad. Sci. USA 2001, 98, 820–822. [Google Scholar] [CrossRef] [PubMed]
- McLaren, A.D.; Peterson, G.H.; Barshad, I. The Adsorption and Reactions of Enzymes and Proteins on Clay Minerals: IV. Kaolinite and Montmorillonite. Soil Sci. Soc. Am. J. 1958, 22, 239–244. [Google Scholar] [CrossRef]
- Sieskind, O. Étude des complexes d’adsorption formés entre la montmorillonite-H et certains acides aminés. Isothermes d’adsorption à pH 2 et à 200 °C. Compt. Rend. Acad. Sci. Paris 1960, 250, 2228–2230. [Google Scholar]
- Cloos, P.; Calicis, B.; Fripiat, J.J.; Makay, K. Adsorption of amino-acids and peptides by montmorillonite. I. Chemical and X-ray diffraction studies. In Proceedings of the International Clay Conference, Jerusalem, Israel, 20–24 June 1966; pp. 223–232. [Google Scholar]
- Shimoyama, A.; Ponnamperuma, C. Adsorption of Some Amino Acids on Na-Montmorillonite: Implication of the Adsorption for Chemical Evolution. In Biogeochemistryof Amino Acids; Hare, P.E., Hoering, T.C., King, J.K., Eds.; John Wiley & Sons: New York, NY, USA, 1980; pp. 145–151. [Google Scholar]
- Friebele, E.; Shimoyama, A.; Ponnamperuma, C. Adsorption of protein and non-protein amino acids on a clay mineral: A possible role of selection in chemical evolution. J. Mol. Evol. 1980, 16, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Bada, J.L.; Miller, S.L. Ammonium ion concentration in the primitive ocean. Science 1968, 159, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Greenland, D.J.; Laby, R.H.; Quirk, J.P. Adsorption of Glycine and Its Di-, Tri-, and Tetra-Peptides by Montmorillonite. Trans. Farad. Soc. 1962, 58, 829–841. [Google Scholar] [CrossRef]
- Greenland, D.J.; Laby, R.H.; Quirk, J.P. Adsorption of Amino-Acids and Peptides by Montmorillinite and Illite. Trans. Farad. Soc. 1965, 61, 2024–2035. [Google Scholar] [CrossRef]
- Lawless, J.G.; Banin, A.; Church, F.M.; Mazzurco, J.; Huff, R.; Kao, J.; Cook, A.; Lowe, T.; Orenberg, J.B. pH Profile of the Adsorption of Nucleotides onto Montmorillonite I. Selected Homoionic Clays. Orig. Life 1985, 15, 77–87. [Google Scholar] [CrossRef]
- Banin, A.; Lawless, J.G.; Mazzurco, J.; Church, F.M.; Margulies, L.; Orenberg, J.B. pH Profile of the Adsorption of Nucleotides onto Montmorillonite II Adsorption and Desorption of 5′-AMP in Iron-Calcium Montmorillonite System. Orig. Life 1985, 15, 89–101. [Google Scholar] [CrossRef]
- Winter, D.; Zubay, G. Binding of Adenine and Adenine-Related Compounds to the Clay Montmorillonite and the Mineral Hydroxylapatite. Orig. Life Evol. Biosph. 1995, 25, 61–81. [Google Scholar] [CrossRef]
- Hashizume, H.; van der Gaast, S.; Theng, B.K.G. Adsorption of Adenine, Cytosine, Uracil, Ribose, and Phosphate by Mg-exchanged Montmorillonite. Clay Miner. 2010, 45, 413–419. [Google Scholar] [CrossRef]
- Hashizume, H.; Theng, B.K.G. Adenine, Adenosine, Ribose and 5′-AMP Adsorption to Allophane. Clays Clay Miner. 2007, 55, 599–605. [Google Scholar] [CrossRef]
- Theng, B.K.G.; Russell, M.; Churchman, G.J.; Parfitt, R.L. Surface Properties of Allophane, Halloysite, and Imogolite. Clays Clay Miner. 1982, 30, 143–149. [Google Scholar] [CrossRef]
- Kalra, S.; Pant, C.K.; Pathak, H.D.; Mehta, M.S. Adsoprtion of glycine and alanine on montmorillonite with or without coordinated divalent cations. Ind. J. Biochem. Biophys. 2000, 37, 341–346. [Google Scholar]
- Carneiro, C.E.A.; Berndt, G.; de Junior, I.G.S.; de Souza, C.M.D.; Paesano, A., Jr.; da Costa, A.C.S.; di Mauro, E.; de Santana, H.; Zaia, C.T.B.V.; Zaia, D.A.M. Adsorption of Adenine, Cytosine, Thymine, and Uracil on Sulfide-Modified Montmorillonite: FT-IR, Mössbauer and EPR Spectroscopy and X-Ray Diffractometry Studies. Orig. Life Evol. Biosph. 2011, 41, 453–468. [Google Scholar] [CrossRef]
- Gururani, K.; Pant, C.K.; Pathak, H.D. Surface Interaction of Adenine on Montmorillonite Clay In Presence and Absence of Divalent Cations In Relevance To Chemical Evolution. Int. J. Sci. Tech. Res. 2012, 1, 106–109. [Google Scholar]
- Jaber, M.; Georgelin, T.; Bazzi, H.; Costa-Torro, F.; Lambert, J.-F.; Bolbach, G.; Clodic, G. Selectivities in Adsorption and Peptidic Condensation in the (Arginine and Glutamic Acid)/Montmorillonite Clay System. J. Phys. Chem. C 2014, 118, 25447–25455. [Google Scholar] [CrossRef]
- Michalkova, A.; Robinson, T.L.; Leszezynski, J. Adsorption of Thymine and Uracil on 1:1 Clay Mineral Surfaces: Comprehensive Ab Initio Study on Influence of Sodium Cation and Water. Phys. Chem. Chem. Phys. 2011, 13, 7862–7881. [Google Scholar] [CrossRef]
- Mignon, P.; Ugliengo, P.; Sodupe, M. Theoretical Study of the Adsorption of RNA/DNA Bases on the External Surfaces of Na+-Montmorillonite. J. Phys. Chem. C 2009, 113, 13741–13749. [Google Scholar] [CrossRef]
- Mignon, P.; Sodupe, M. Structural Behaviors of Cytosine into the Hydrated Interlayer of Na+-Montmorillonite Clay. An ab Initio Molecular Dynamics Study. J. Phys. Chem. C 2013, 117, 26179–26189. [Google Scholar] [CrossRef]
- Mignon, P.; Sodupe, M. Theoretical study of the adsorption of DNA bases on the acidic external surface of montmorillonite. Phys. Chem. Chem. Phys. 2012, 14, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Roa, E.; Huertas, F.J.; Hernández-Laguna, A.; Sainz-Díaz, C.I. A DFT study of the adsorption of glycine in the interlayer space of montmorillonite. Phys. Chem. Chem. Phys. 2017, 19, 14961–14971. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.E.; Huertas, F.J. Adsorption of glycine on montmorillonite in aqueous solutions. Appl. Clay Sci. 2013, 80–81, 10–17. [Google Scholar] [CrossRef]
- Hashizume, H. Adsorption of Some Amino Acids by Chrysotile. Viva Origino 2007, 35, 60–65. [Google Scholar]
- Hashizume, H.; Theng, B.K.G. Adsorption of DL-Alanine by Allophane: Effect of pH and Unit Particle Aggregation. Clay Miner. 1999, 34, 233–238. [Google Scholar] [CrossRef]
- Hashizume, H.; Theng, B.K.G. Adsorption of L-Alanine Monomer, Dimer, Tetramer, Pentamer by Some Allophanes. In Proceedings of the 9th International Symposium Water-Rock Interaction, Taupo, New Zealand, 30 March–3 April 1998; pp. 105–107. [Google Scholar]
- Gabel, N.W.; Ponnamperuma, C. Model for Origin of Monosaccharides. Nature 1967, 216, 453–455. [Google Scholar] [CrossRef]
- Saladino, R.; Neri, V.; Crestini, C. Role of Clays in the Prebiotic Synthesis of Sugar Derivatives from Formamide. Philos. Mag. 2010, 90, 2329–2337. [Google Scholar] [CrossRef]
- Nakazawa, H.; Yamada, H.; Hashizume, H. Origin of Life in the Earth’s Crust, a Hypothesis: Probable Chemical Evolution Synchronized with the Plate Tectonics of the Early Earth. Viva Origino 1993, 21, 213–222, (In Japanese with English abstract). [Google Scholar]
- Ohara, S.; Kakegawa, T.; Nakazawa, H. Pressure Effects on the Abiotic Polymerization of Glycine. Orig. Life Evol. Biosph. 2007, 37, 215–223. [Google Scholar] [CrossRef]
- Paecht-Horowitz, M.; Berger, J.; Katchalsky, A. Prebiotic Synthesis of Polypeptides by Heterogeneous Polycondensation of Amino-acid Adenylates. Nature 1970, 228, 636–639. [Google Scholar] [CrossRef]
- Paecht-Horowitz, M. Inorganic Clays as Possible Prebiotic Peptide Templates. Isr. J. Chem. 1973, 11, 369–378. [Google Scholar] [CrossRef]
- Paecht-Horowitz, M. The possible role of clays in prebiotic peptide synthesis. Orig. Life 1974, 5, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Blank, J.G.; Miller, G.H.; Ahrens, M.J.; Winans, R.E. Experimental Shock Chemistry of Aqueous Amino Acid Solution and the Cometary Delivery of Prebiotic Compounds. Orig. Life Evol. Biosph. 2001, 31, 15–51. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Ertem, G. Montmorillonite Catalysis of RNA Oligomer Formation in Aqueous Solution. A Model for the Prebiotic Formation of RNA. J. Am. Chem. Soc. 1993, 115, 12270–12275. [Google Scholar] [CrossRef]
- Ferris, J.P.; Hill, A.R., Jr.; Liu, R.; Orgel, L.E. Synthesis of Long Prebiotic Oligomers on Mineral Surfaces. Nature 1996, 381, 59–61. [Google Scholar] [CrossRef]
- Joshi, P.C.; Aldersley, M.F.; Delano, J.W.; Ferris, J.P. Mechanism of Montmorillonite Catalysis in the Formation of RNA Oligomers. J. Am. Chem. Soc. 2009, 131, 13369–13374. [Google Scholar] [CrossRef]
- Ertem, G.; Ferris, J.P. Sequence- and regio-selectivity in the montmorillonite-catalyzed synthesis of RNA. Orig. Life Evol. Biosph. 2000, 30, 411–422. [Google Scholar] [CrossRef]
- Ertem, G.; Prabahar, K.J.; Joshi, P.C.; Ferris, J.P. Bridging the prebiotic and RNA worlds: Prebiotic RNA synthesis on clay. J. Biomol. Struct. Dyn. 2000, 17 (Suppl 1), 207–210. [Google Scholar] [CrossRef]
- Ferris, J.P.; Joshi, P.C.; Ertem, G.; Wang, K.-J. Catalysis in prebiotic synthesis: Montmorillonite catalysis of RNA formation. In Proceedings of the Abstracts of Papers, 221st ACS National Meeting, San Diego, CA, USA, 1–5 April 2001; p. GEOC-156. [Google Scholar]
- Ferris, J.P. Montmorillonite Catalysis of 30–50 Mer Oligonucleotides: Laboratory Demonstration of Potential Steps in the Origin of the RNA World. Orig. Life Evol. Biosph. 2002, 32, 311–332. [Google Scholar] [CrossRef]
- Huang, W.; Ferris, J.P. Synthesis of 35–40 mers of RNA oligomers from unblocked monomers. A simple approach to the RNA world. Chem. Commun. 2003, 1458–1459. [Google Scholar] [CrossRef]
- Miyakawa, S.; Joshi, P.C.; Gaffey, M.J.; Gonzalez-Toril, E.; Hyland, C.; Ross, T.; Rybij, K.; Ferris, J.P. Studies in the Mineral and Salt-Catalyzed Formation of RNA Oligomers. Orig. Life Evol. Biosph. 2006, 36, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Aldersley, M.F.; Joshi, P.C. RNA dimer synthesis using montmorillonite as a catalyst: The role of surface layer charge. Appl. Clay Sci. 2013, 83–84, 77–82. [Google Scholar] [CrossRef]
- Joshi, P.C.; Dubey, K.; Aldersley Michael, F.; Sausville, M. Clay catalyzed RNA synthesis under Martian conditions: Application for Mars return samples. Biochem. Biophys. Res. Commun. 2015, 462, 99–104. [Google Scholar] [CrossRef]
- Namani, T.; Snyder, S.; Eagan, J.M.; Bevilacqua, P.C.; Wesdemiotis, C.; Sahai, N. Amino Acid Specific Nonenzymatic Montmorillonite-Promoted RNA Polymerization. Chem. Systems Chem. 2021, 3, e2000060. [Google Scholar] [CrossRef]
- Schneider, J. A model for non-chemical form of life: Crystalline physiology. Orig. Life 1977, 8, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Cairns-Smith, A.G. Seven Clues to the Origin of Life: A Scientific Detective Story; Cambridge University Press: Cambridge, UK; New York, NY, USA, 1991. [Google Scholar]
- Bullard, T.; Freudenthal, J.; Avagyan, S.; Kahr, B. Test of Cairns-Smith’s ‘crystals-as-genes’ hypothesis. Faraday Discuss. 2007, 136, 231–245. [Google Scholar] [CrossRef]
- Siffert, B. Quelques Réactions de la Silice en Solution: La Formation des Argiles; Service de la carte géologique d’Alsace et de Lorraine: Strasbourg, France, 1962; Volume 21, p. 102. [Google Scholar]
- Small, J.S.; Hamilton, D.L.; Habesch, S. Experimental simulations of clay precipitation within reservoir sandstones. 2. Mechanism of illite formation and controls on morphology. J. Sediment. Petrol. 1992, 62, 520–529. [Google Scholar] [CrossRef]
- Small, J.S.; Manning, D.A.C. Laboratory reproduction of morphological variation in petroleum reservoir clays: Monitoring of fluid composition during illite precipitation. In Geochemistry of Clay Pore Fluid Interactions; Manning, D.A.C., Hall, P.L., Hughess, C.R., Eds.; Mineralogical Society/Chapman and Hall: London, UK, 1993; pp. 181–212. [Google Scholar]
- Hartman, H. Photosynthesis and the origin of life. Orig. Life Evol. Biosph. 1998, 28, 515–521. [Google Scholar] [CrossRef]
- Cape, J.L.; Monnard, P.-A.; Boncella, J.M. Prebiotically relevant mixed fatty acid vesicles support anionic solute encapsulation and photochemically catalyzed trans-membrane charge transport. Chem. Sci. 2011, 2, 661–671. [Google Scholar] [CrossRef]
- Deamer, D.W.; Pashley, R.M. Amphiphilic components of the Murchison carbonaceous chondrite: Surface properties and membrane formation. Orig. Life Evol. Biosph. 1989, 19, 21–38. [Google Scholar] [CrossRef]
- Smith, J.V. Biochemical evolution. I. Polymerization on internal, organophilic silica surfaces of dealuminated zeolites and feldspars. Proc. Nat. Acad. Sci. USA 1998, 95, 3370–3375. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.V.; Arnold, F.P., Jr.; Parsons, I.; Lee, M.R. Biochemical evolution III: Polymerization on organophilic silica-rich surfaces, crystal-chemical modeling, formation of first cells, and geological clues. Proc. Nat. Acad. Sci. USA 1999, 96, 3479–3485. [Google Scholar] [CrossRef] [PubMed]
- Brasier, M.D.; Matthewman, R.; McMahon, S.; Wacey, D. Pumice as a remarkable substrate for the origin of life. Astrobiology 2011, 11, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Hansma, H.G. Possible origin of life between mica sheets. J. Theor. Biol. 2010, 266, 175–188. [Google Scholar] [CrossRef]
- Subramanian, A.B.; Wan, J.; Gopinath, A.; Stone, H.A. Semi-Permeable Vesicles Composed of Natural Clay. Soft Matter 2011, 7, 2600–2612. [Google Scholar] [CrossRef]
- Hanczyc, M.M.; Fujikawa, S.M.; Szostak, J.W. Experimental models of primitive cellular compartments: Encapsulation, growth, and division. Science 2003, 302, 618–622. [Google Scholar] [CrossRef]
- Hanczyc, M.M.; Mansy, S.S.; Szostak, J.W. Mineral Surface Directed Membrane Assembly. Orig. Life Evol. Biosph. 2007, 37, 67–82. [Google Scholar] [CrossRef]
- Chatzitheodoridis, E.; Haigh, S.; Lyon, I. A conspicuous clay ovoid in Nakhla: Evidence for subsurface hydrothermal alteration on Mars with implications for astrobiology. Astrobiology 2014, 14, 651–693. [Google Scholar] [CrossRef]
- Schrödinger, E. What Is Life?: The Physical Aspect of the Living Cell; Cambridge University Press: Cambridge, UK, 1944; p. 91. [Google Scholar]
- Horowitz, N.H. On the Evolution of Biochemical Syntheses. Proc. Nat. Acad. Sci. USA 1945, 31, 153–157. [Google Scholar] [CrossRef]
- Granick, S. Speculations on the origins and evolution of photosynthesis. Ann. N. Y. Acad. Sci. 1957, 69, 292–308. [Google Scholar] [CrossRef]
- Hartman, H. Speculations on the evolution of the genetic code. Orig. Life 1975, 6, 423–427. [Google Scholar] [CrossRef]
- Hartman, H.; Smith, T.F. Origin of the Genetic Code Is Found at the Transition between a Thioester World of Peptides and the Phosphoester World of Polynucleotides. Life 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Cairns-Smith, A.G.; Hartman, H. (Eds.) Clays as catalysts. In Clay Minerals And The Origin Of Life; Cambridge University Press: Cambridge, UK, 1986; pp. 130–138. [Google Scholar]
- Metzler, D.E.; Snell, E.E. Deamination of serine. I. Catalytic deamination of serine and cysteine by pyridoxal and metal salts. J. Biol. Chem. 1952, 198, 353–361. [Google Scholar] [CrossRef]
- Mortland, M.M. Deamination of glutamic acid by pyridoxal phosphate-Cu2+-smectite catalysts. J. Mol. Catal. 1984, 27, 143–155. [Google Scholar] [CrossRef]
- Cairns-Smith, A.G. Genes made of clay. New Sci. 1974, 64, 274–276. [Google Scholar]
- Cairns-Smith, A.G. The first organisms. Sci. Am. 1985, 252, 74–82. [Google Scholar] [CrossRef]
- Boyd, S.A.; Mortland, M.M. Manipulating the activity of immobilized enzymes with different organo-smectite complexes. Experientia 1985, 12, 1564–1566. [Google Scholar] [CrossRef]
- Boyd, S.A.; Mortland, M.M. Selective effects of smectite-organic complexes on the activities of immobilized enzymes. J. Mol. Catal. 1986, 34, 1–8. [Google Scholar] [CrossRef]
- Siffert, B.; Naidja, A. Décarboxylation catalytique de l’acide oxaloacétique en présence de montmorillonite. Clay Miner. 1987, 22, 435–446. [Google Scholar] [CrossRef]
- Naidja, A.; Siffert, B. Oxidative decarboxylation of isocitric acid in the presence of montmorillonite. Clay Miner. 1990, 25, 27–37. [Google Scholar] [CrossRef]
- Mortland, M.M.; Lawless, J.G.; Hartman, H.; Frankel, R. Smectite interactions with flavomononucleotide. Clays Clay Miner. 1984, 32, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Hartman, H. Mars, Clays and the origins of life. In Proceedings of the Exobiology and Future Mars Missions: A NASA Symposium Held in Sunnyvale, Sunnyvale, CA, USA, 23–25 March 1988; pp. 30–31. [Google Scholar]
- Hartman, H.; Sposito, G.; Yang, A.; Manne, S.; Gould, S.A.C.; Hansma, P.K. Molecular-scale imaging of clay mineral surfaces with the atomic microscope. Clays Clay Miner. 1990, 38, 337–342. [Google Scholar] [CrossRef]
- Siffert, B. The role of organic complexing agents. In Clay Minerals and the Origin of Life; Cairns-Smith, A.G., Hartman, H., Eds.; Cambridge University Press: Cambridge, UK, 1986; pp. 75–78. [Google Scholar]
- Barshad, I. Soil Development. In Chemistry of the Soil; Bear, F.E., Ed.; Reinhold Publishing Corp.: New York, NY, USA, 1955; pp. 1–52. [Google Scholar]
- Vogels, R.J.M.J.; Kerkhoffs, M.J.H.V.; Geus, J.W. Non-hydrothermal synthesis, characterisation and catalytic properties of saponite clays. Stud. Surf. Sci. Catal. 1995, 91, 1153–1161. [Google Scholar]
- Guzman, M.I. Abiotic Photosynthesis: From Prebiotic Chemistry to Metabolism. In Origins of Life: The Primal Self-Organization; Egel, R., Lankenau, D.-H., Mulkidjanian, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 85–105. [Google Scholar]
- Losa-Adams, E.; Gil-Lozano, C.; Fairén, A.G.; Bishop, J.L.; Rampe, E.B.; Gago-Duport, L. Long-lasting habitable periods in Gale crater constrained by glauconitic clays. Nat. Astron. 2021, 5, 936–942. [Google Scholar] [CrossRef]

















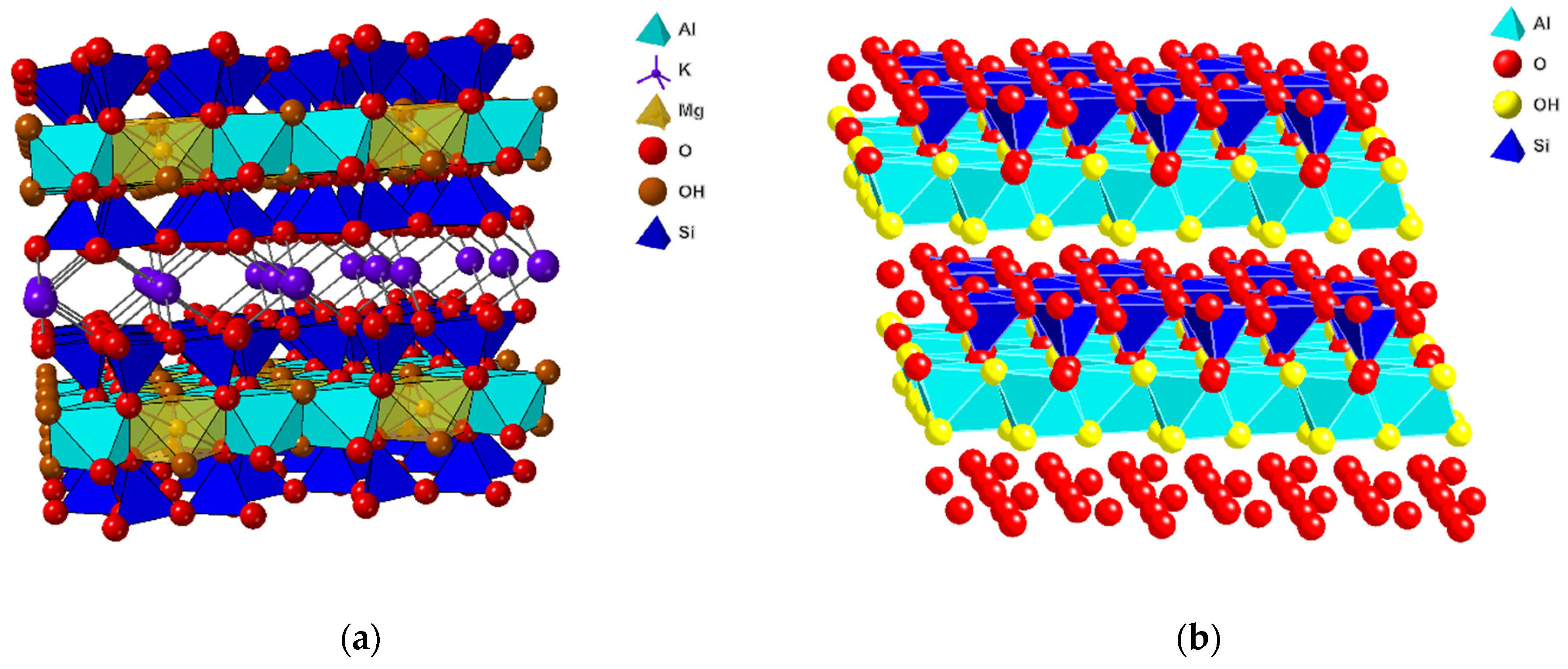{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clay Mineral | Type | Chemical Composition |
|---|---|---|
| Allophane | Short-range-order clay | (Al2O3)(SiO2)1.3–2·2.5–3H2O |
| Kaolinite | 1:1 clays, dioctahedral | Al2Si2O5(OH)4 |
| Lizardite | 1:1 serpentine, trioctahedral | Mg3Si2O5(OH)4 |
| Antigorite | 1:1 serpentine, trioctahedral | Mg3Si2O5(OH)4 |
| Chrysotile | 1:1 serpentine, trioctahedral | Mg3Si2O5(OH)4 |
| Talc | 2:1 clay, trioctahedral | Mg3Si4O10(OH)2 |
| Montmorillonite | 2:1 clays, dioctahedral | (M+y.nH2O)(Al3+2-yMg2+y)Si4+4O10(OH)2 |
| Beidellite | 2:1 clays, dioctahedral | (M+x.nH2O)Al3+2(Si4+4-xAl3+x)O10(OH)2 |
| Nontronite | 2:1 clays, dioctahedral | (M+x.nH2O)Fe3+2(Si4+4-xAl3+x)O10(OH)2 |
| Hectorite | 2:1 clays, trioctahedral | (M+y.nH2O)(Mg2+3-yLi+y)(Si4+4O10(OH)2 |
| Saponite | 2:1 clays, trioctahedral | (M+x.nH2O)Mg2+3(Si4+4-xAl3+x)O10(OH)2 |
| Sauconite | 2:1 clays, trioctahedral | (M+x.nH2O)Zn2+3(Si4+4-xAl3+x)O10(OH)2 |
| Stevensite | 2:1 clays, trioctahedral | (Ca,Na)xMg3-x(Si4O10)(OH)2 |
| Vermiculite | 2:1 clays, trioctahedral | Mg0.7(Mg,Fe,Al)6(Si,Al)8O20(OH)4.8H2O |
| Corrensite | 1:1 regular interstratified 1 | (Mg,Fe)9((Si,Al)8O20)(OH)10·nH2O |
| Chlorite | 2:1 clays, interlayer octahedral sheet | ([Mg2+Fe2+]5Al3+)(Si4+3Al3)O10(OH)8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloprogge, J.T.; Hartman, H. Clays and the Origin of Life: The Experiments. Life 2022, 12, 259. https://doi.org/10.3390/life12020259
Kloprogge JT, Hartman H. Clays and the Origin of Life: The Experiments. Life. 2022; 12(2):259. https://doi.org/10.3390/life12020259
Chicago/Turabian StyleKloprogge, Jacob Teunis (Theo), and Hyman Hartman. 2022. "Clays and the Origin of Life: The Experiments" Life 12, no. 2: 259. https://doi.org/10.3390/life12020259
APA StyleKloprogge, J. T., & Hartman, H. (2022). Clays and the Origin of Life: The Experiments. Life, 12(2), 259. https://doi.org/10.3390/life12020259