
Membrane Structure Obtained in an Experimental Evolution Process



Abstract
:1. Introduction
2. Computational Methods
2.1. Simulation Systems
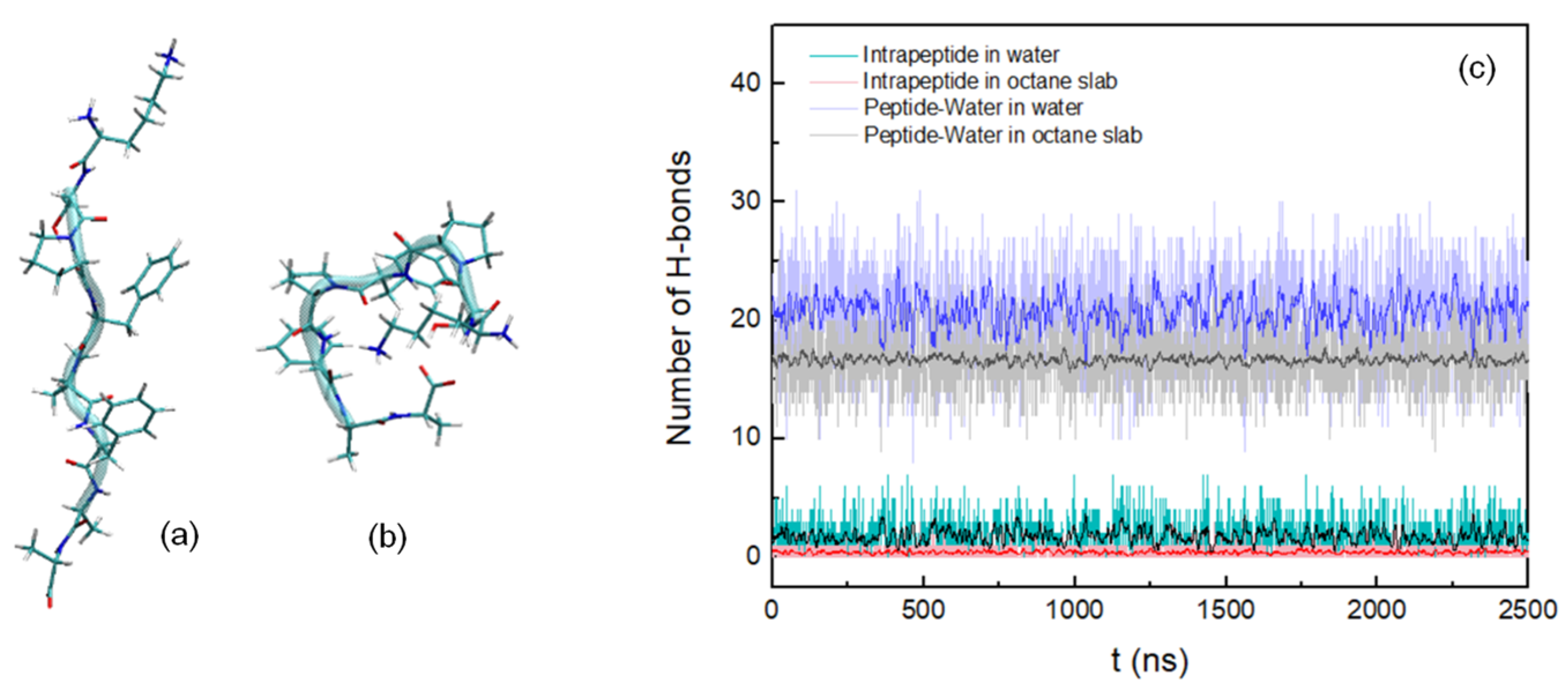
2.1.1. Peptide in Explicit Solvent
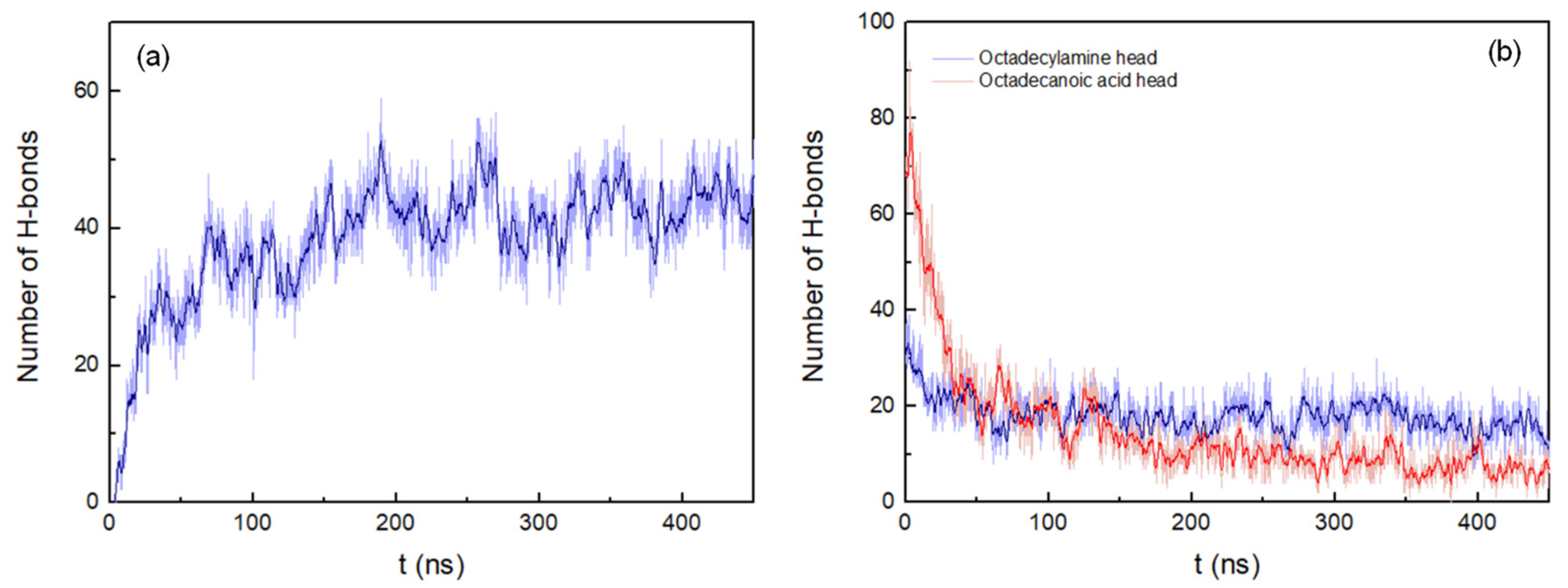
2.1.2. Peptides in Octadecylamine/Octadecanoic Acid Bilayer
2.2. Simulation Setup
3. Results
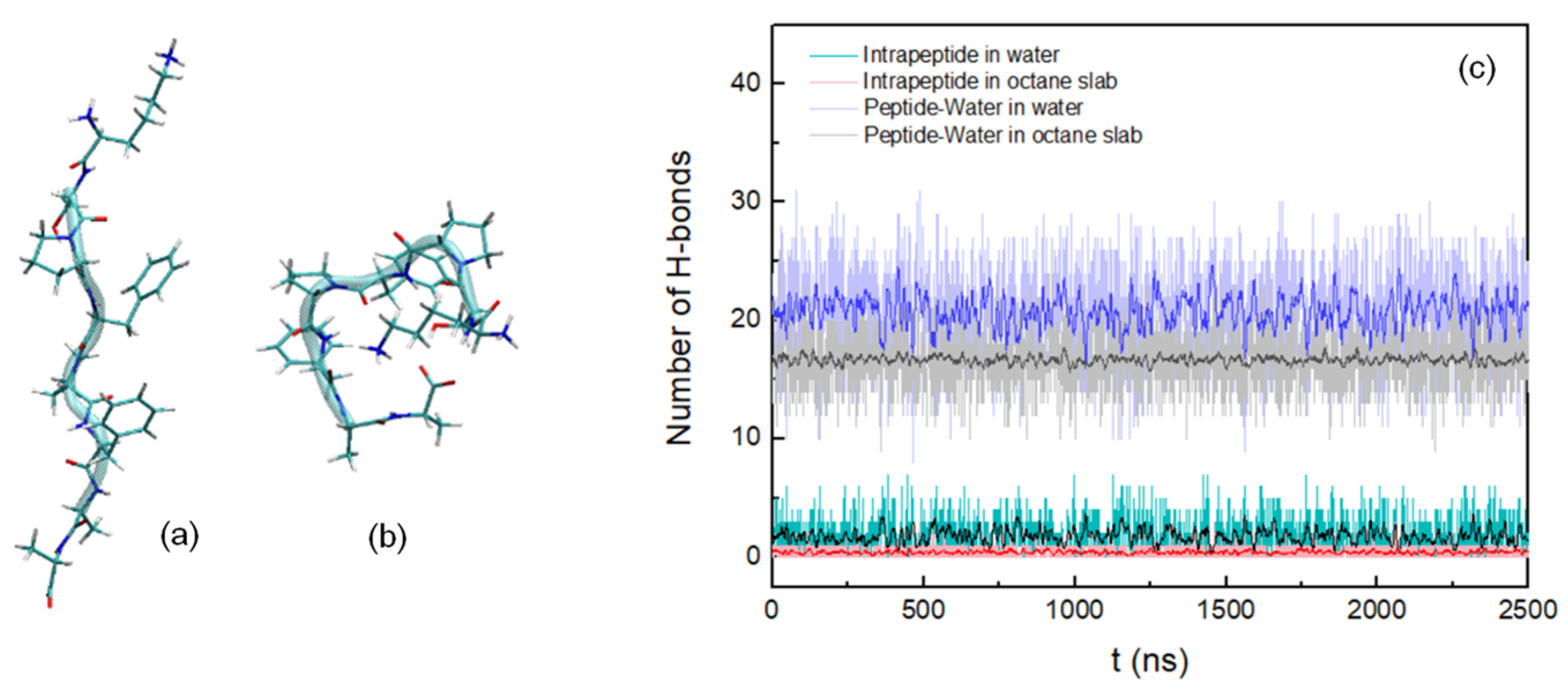
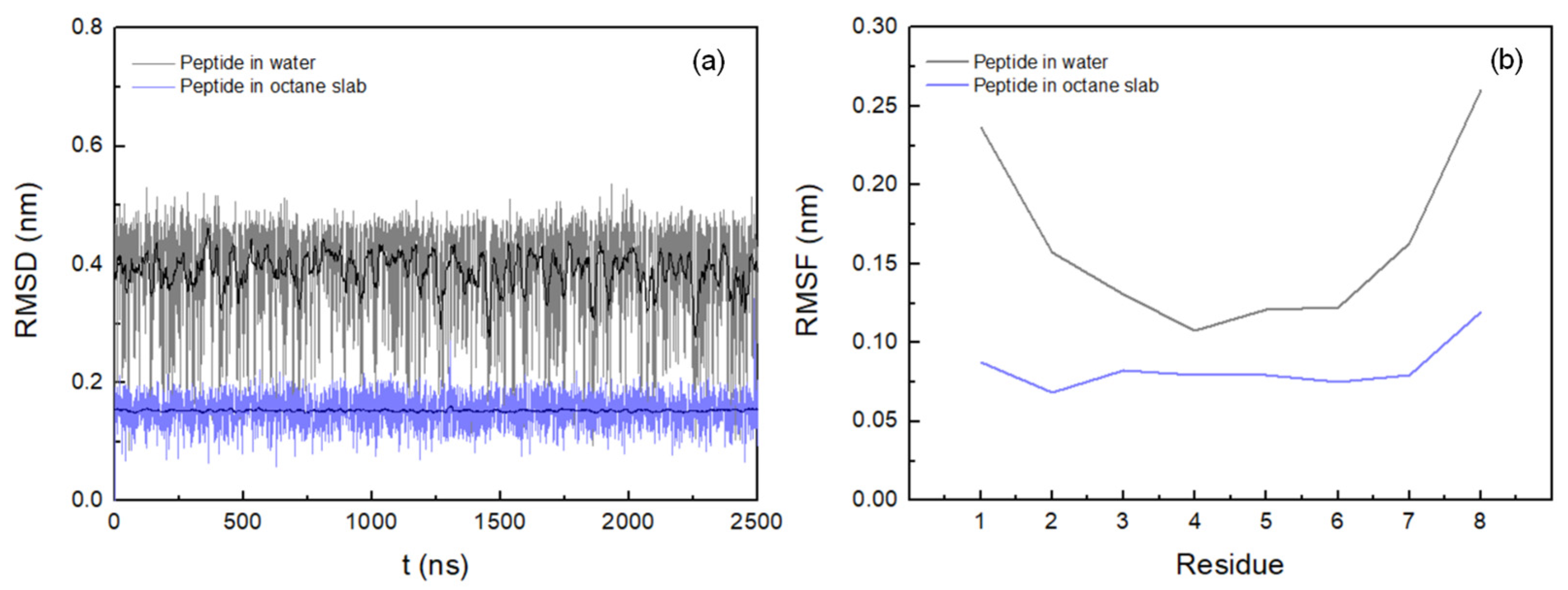
3.1. Peptide Conformation into Biphasic Membrane Mimetic
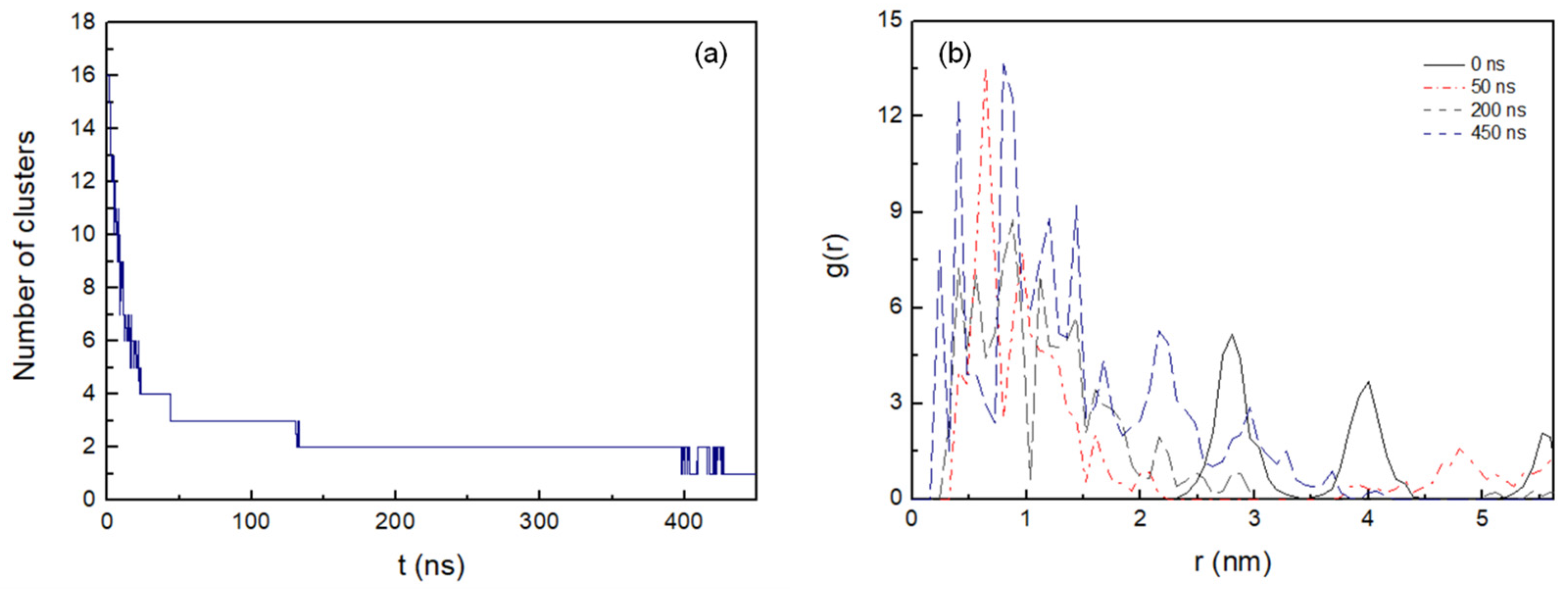
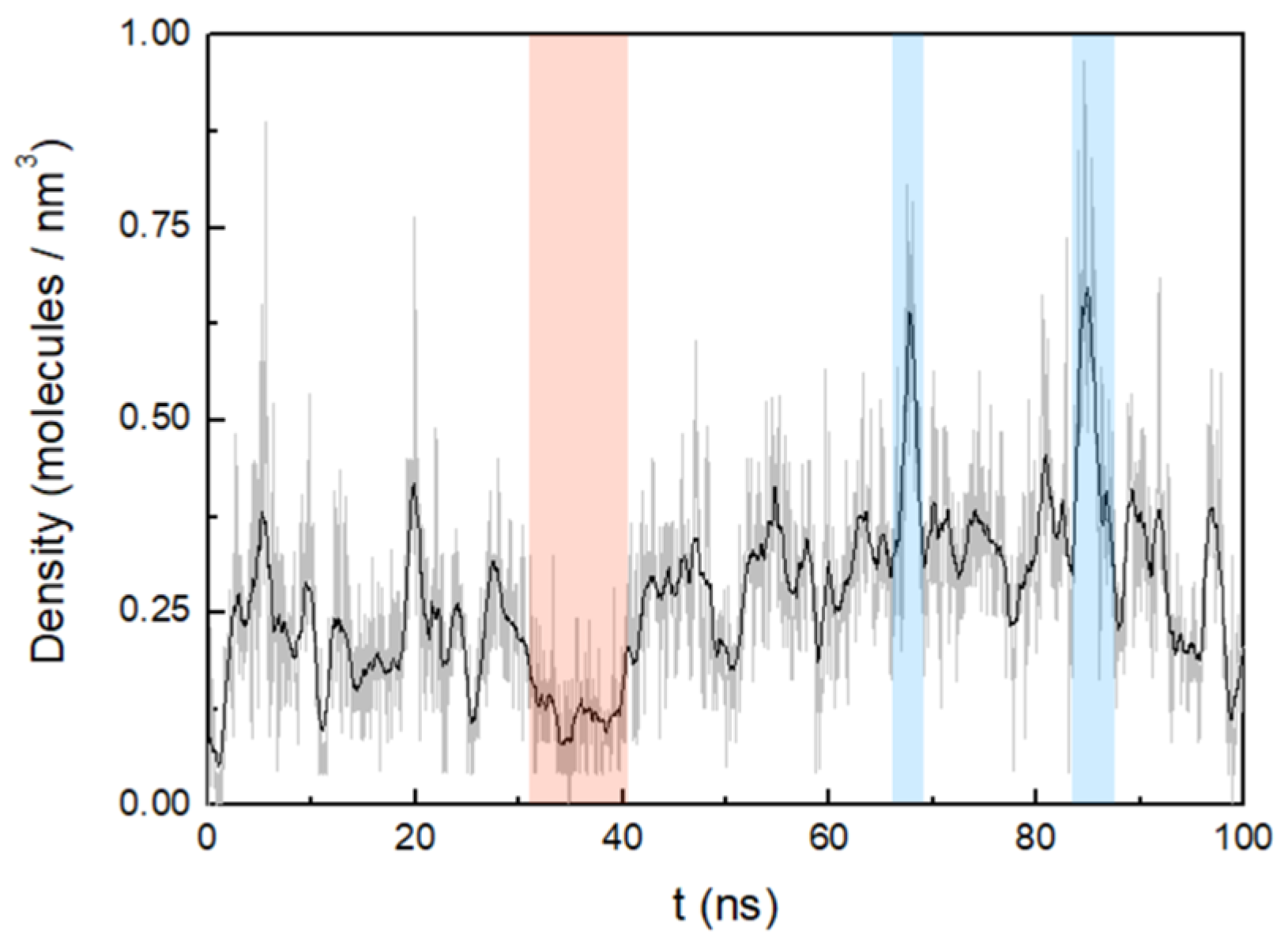
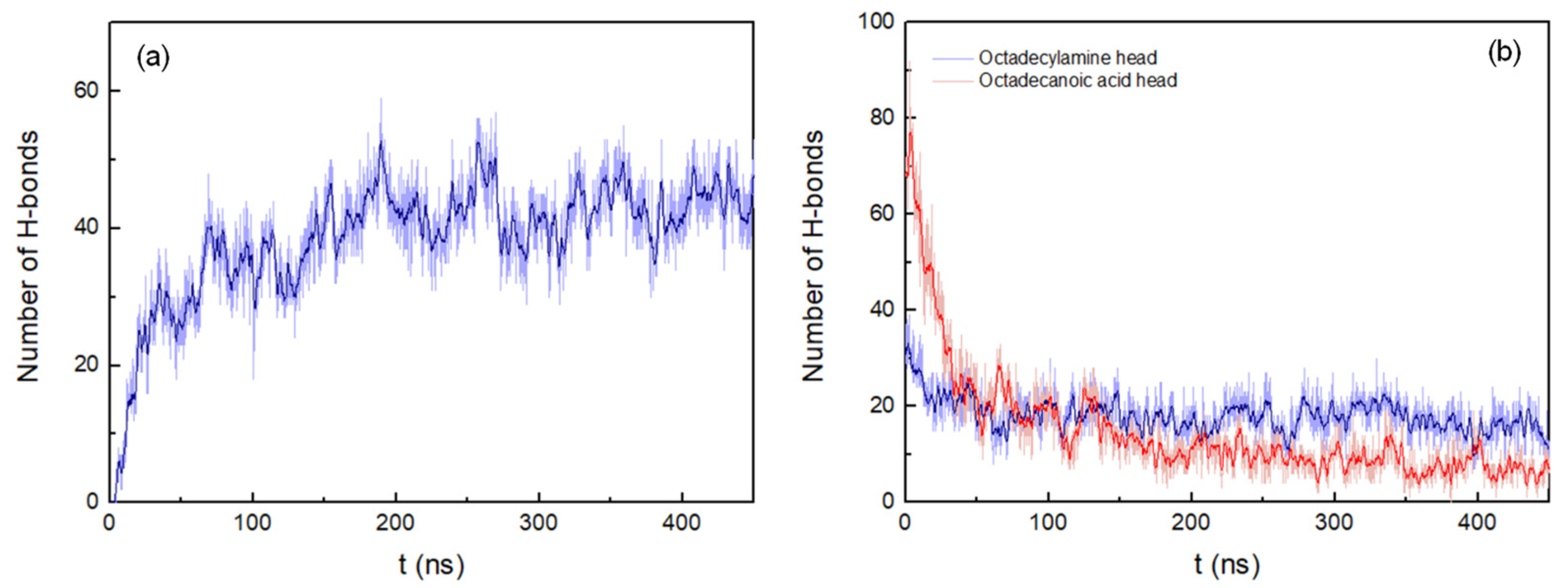
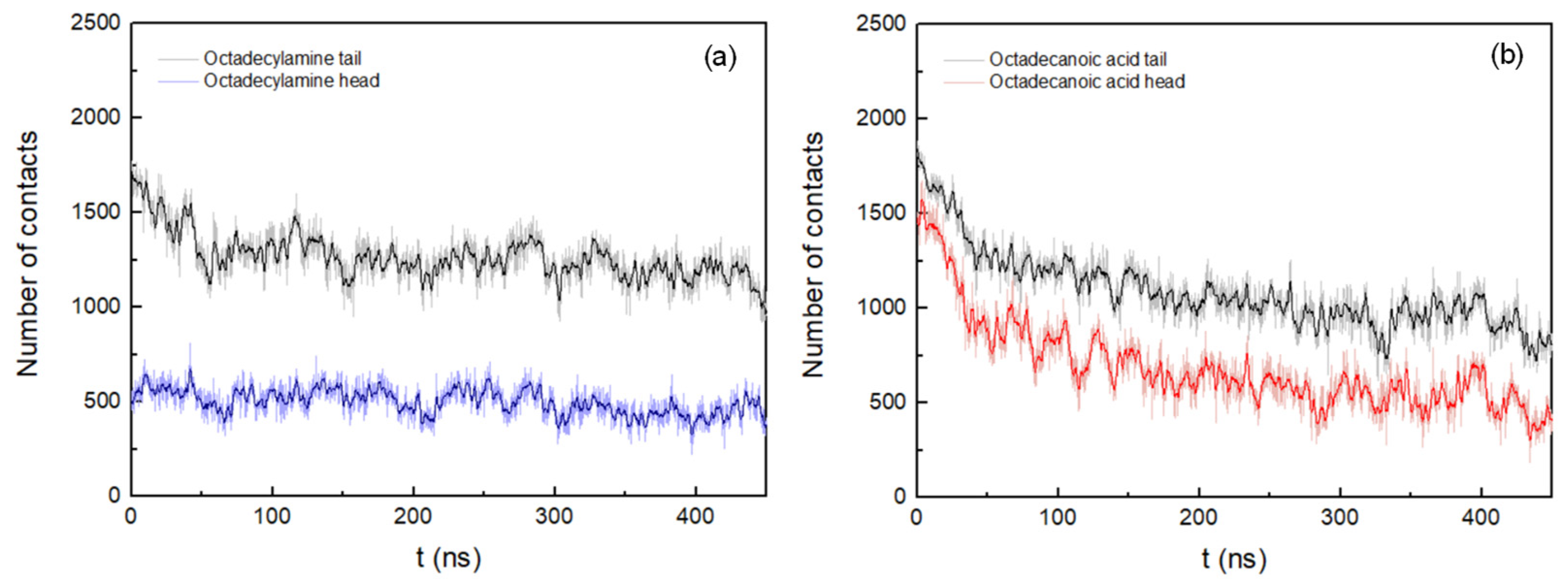
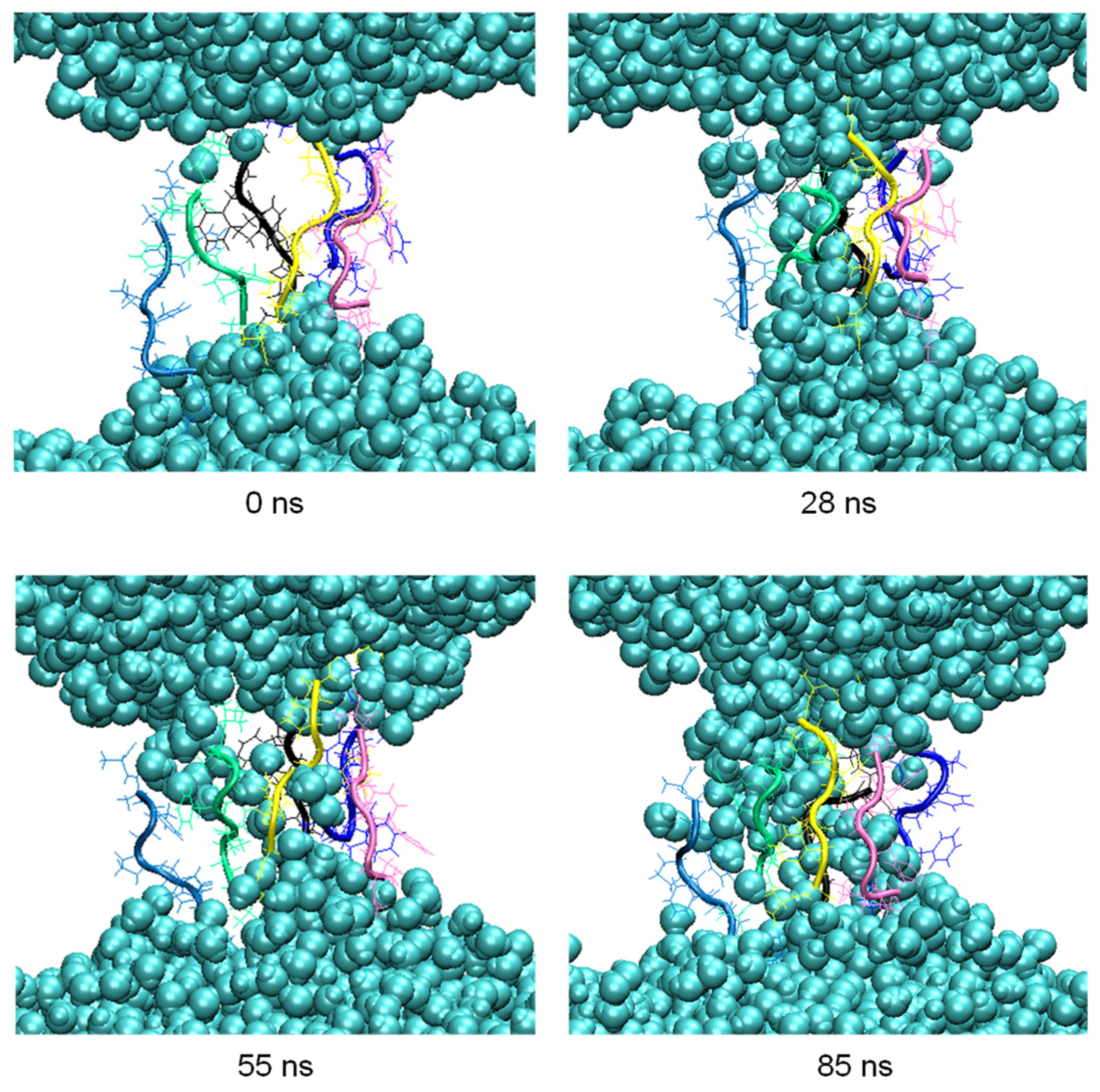
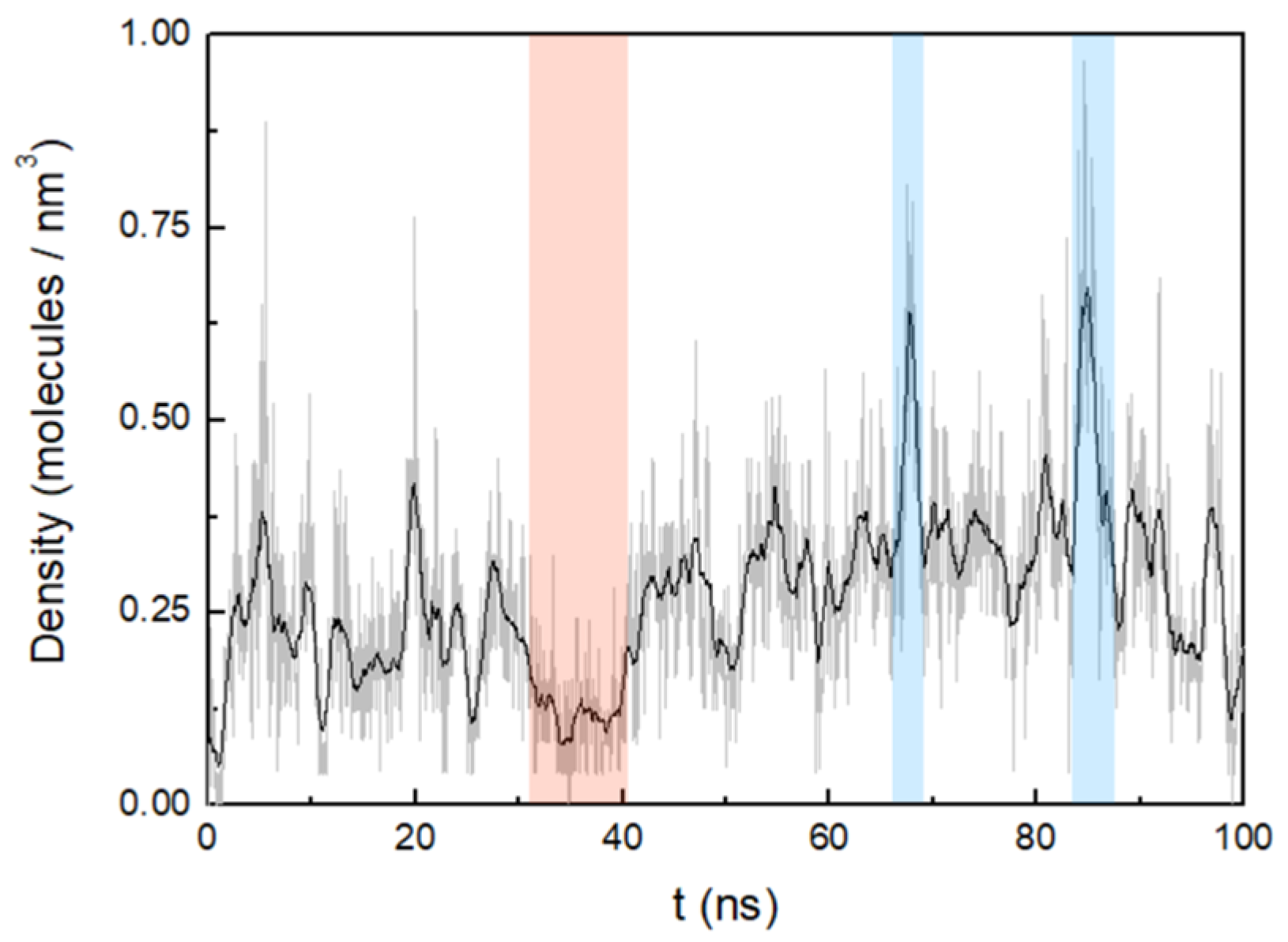
3.2. Cluster Process and Pore Formation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schrum, J.P.; Zhu, T.F.; Szostak, J.W. The origins of cellular life. Cold Spring Harb. Perspect. Biol. 2010, 2, a002212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deamer, D. The role of lipid membranes in life’s origin. Life 2017, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.A.; Szostak, J.W. A kinetic study of the growth of fatty acid vesicles. Biophys. J. 2004, 87, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansy, S.S. Membrane transport in primitive cells. Cold Spring Harb. Perspect. Biol. 2010, 2, a002188. [Google Scholar] [CrossRef] [Green Version]
- Toparlak, Ö.D.; Wang, A.; Mansy, S.S. Population-level membrane diversity triggers growth and division of protocells. JACS Au 2021, 1, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.-A.; Deamer, D.W. Membrane self-assembly processes: Steps toward the first cellular life. Anat. Rec. 2002, 268, 196–207. [Google Scholar] [CrossRef]
- Sacerdote, M.G.; Szostak, J.W. Semipermeable lipid bilayers exhibit diastereoselectivity favoring ribose. Proc. Natl. Acad. Sci. USA 2005, 102, 6004–6008. [Google Scholar] [CrossRef] [Green Version]
- Deamer, D.W. Boundary structures are formed by organic components of the Murchison carbonaceous chondrite. Nature 1985, 317, 792–794. [Google Scholar] [CrossRef]
- Rushdi, A.I.; Simoneit, B.R. Lipid formation by aqueous Fischer-Tropsch-type synthesis over a temperature range of 100 to 400 degrees C. Orig. Life Evol. Biosph. 2001, 31, 103–118. [Google Scholar] [CrossRef]
- Schreiber, U.; Mayer, C.; Schmitz, O.J.; Rosendahl, P.; Bronja, A.; Greule, M.; Keppler, F.; Mulder, I.; Sattler, T.; Schöler, H.F. Organic compounds in fluid inclusions of Archean quartz—Analogues of prebiotic chemistry on early Earth. PLoS ONE 2017, 12, e0177570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, C.; Mautner, M.; Deamer, D.W. Self-assembled vesicles of monocarboxylic acids and alcohols: Conditions for stability and for the encapsulation of biopolymers. Biochim. Biophys. Acta Biomembr. 2002, 1559, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.F.; Rammu, H.; Zheludev, I.N.; Hartley, A.M.; Marechal, A.; Lane, N. Promotion of protocell self-assembly from mixed amphiphiles at the origin of life. Nat. Ecol. Evol. 2019, 3, 1705–1714. [Google Scholar] [CrossRef]
- Maurer, S.E.; Deamer, D.W.; Boncella, J.M.; Monnard, P.-A. Chemical evolution of amphiphiles: Glycerol monoacyl derivatives stabilize plausible prebiotic membranes. Astrobiology 2009, 9, 979–987. [Google Scholar] [CrossRef]
- Groen, J.; Deamer, D.; Kros, A.; Ehrenfreund, P. Polycyclic aromatic hydrocarbons as plausible prebiotic membrane components. Orig. Life Evol. Biosph. 2012, 42, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Namani, T.; Deamer, D.W. Stability of model membranes in extreme environments. Orig. Life Evol. Biosph. 2008, 38, 329–341. [Google Scholar] [CrossRef]
- Damer, B.; Deamer, D. The hot spring hypothesis for an origin of life. Astrobiology 2020, 20, 429–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, A.; Damer, B. Factoring origin of life hypotheses into the search for life in the solar system and beyond. Life 2020, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, U.; Locker-Grütjen, O.; Mayer, C. Hypothesis: Origin of life in deep-reaching tectonic faults. Orig. Life Evol. Biosph. 2012, 42, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Schreiber, U.; Dávila, M.J. Periodic vesicle formation in tectonic fault zones: An ideal scenario for molecular evolution. Orig. Life Evol. Biosph. 2015, 45, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, W.L. Hydrothermal synthesis of amino acids. Geochim. Cosmochim. Acta 1994, 58, 2099–2106. [Google Scholar] [CrossRef]
- Mayer, C.; Schreiber, U.; Dávila, M.J. Selection of prebiotic molecules in amphiphilic environments. Life 2017, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Schreiber, U.; Dávila, M.J.; Schmitz, O.J.; Bronja, A.; Meyer, M.; Klein, J.; Meckelmann, S.W. Molecular evolution in a peptide-vesicle system. Life 2018, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2003. [Google Scholar]
- Abraham, J.M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Intermolecular Forces; Pullmann, B., Ed.; Reidel: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Thøgersen, L.; Schiøt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipot, C.; Pohorille, A. Folding and translocation of the undecamer of poly-L-leucine across the water-hexane interface. A molecular dynamics study. J. Am. Chem. Soc. 1998, 120, 11912–11924. [Google Scholar] [CrossRef] [PubMed]
- Stockner, T.; Ash, W.L.; MacCallum, J.L.; Tieleman, D.P. Direct simulation of transmembrane helix association: Role of asparagines. Biophys. J. 2004, 87, 1650–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hénin, J.; Pohorille, A.; Chipot, C. Insights into the recognition and association of transmembrane α-helices. The free energy of α-helix dimerization in glycophorin A. J. Am. Chem. Soc. 2005, 127, 8478–8484. [Google Scholar] [CrossRef]
- Kulleperuma, K.; Smith, S.M.E.; Morgan, D.; Musset, B.; Holyoake, J.; Chakrabarti, N.; Cherny, V.V.; DeCoursey, T.E.; Pomès, R. Construction and validation of a homology model of the human voltage-gated proton channel hHV1. J. Gen. Physiol. 2013, 141, 445–465. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, E.; Killian, J.A. Snorkeling of lysine side chains in transmembrane helices: How easy can it get? FEBS Lett. 2003, 544, 69–73. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide H-Bonds (hydrated octane slab)/% | |||||||
| S2-P3 | F4-F6 | F6-F8 | |||||
| 87 | 4 | 5 | |||||
| Peptide H-Bonds (water)/% | |||||||
| K1-S2 | K1-A8 | S2-F4 | S2-A8 | F4-F6 | F4-A7 | ||
| 3 | 23 | 23 | 31 | 6 | 4 | ||
| Peptide–Water H-Bonds (hydrated octane slab)/% | |||||||
| K1 | S2 | F4 | A7 | A8 | |||
| 32 | 14 | 3 | 5 | 42 | |||
| Peptide–Water H-Bonds (water)/% | |||||||
| K1 | S2 | P3 | F4 | P5 | F6 | A7 | A8 |
| 26 | 13 | 6 | 5 | 6 | 7 | 9 | 28 |
| Number of Contacts–Octadecylamine (head)/% | |||||||
| K1 | S2 | P3 | F4 | P5 | F6 | A7 | A8 |
| 35 | 9 | 9 | 5 | 5 | 9 | 14 | 14 |
| Number of Contacts–Octadecanoic Acid (head)/% | |||||||
| K1 | S2 | P3 | F4 | P5 | F6 | A7 | A8 |
| 16 | 10 | 14 | 16 | 6 | 17 | 7 | 13 |
| Number of Contacts–Octadecylamine (tail)/% | |||||||
| K1 | S2 | P3 | F4 | P5 | F6 | A7 | A8 |
| 12 | 8 | 12 | 19 | 13 | 18 | 8 | 11 |
| Number of Contacts–Octadecanoic Acid (tail)/% | |||||||
| K1 | S2 | P3 | F4 | P5 | F6 | A7 | A8 |
| 8 | 7 | 13 | 21 | 15 | 20 | 8 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dávila, M.J.; Mayer, C. Membrane Structure Obtained in an Experimental Evolution Process. Life 2022, 12, 145. https://doi.org/10.3390/life12020145
Dávila MJ, Mayer C. Membrane Structure Obtained in an Experimental Evolution Process. Life. 2022; 12(2):145. https://doi.org/10.3390/life12020145
Chicago/Turabian StyleDávila, María J., and Christian Mayer. 2022. "Membrane Structure Obtained in an Experimental Evolution Process" Life 12, no. 2: 145. https://doi.org/10.3390/life12020145
APA StyleDávila, M. J., & Mayer, C. (2022). Membrane Structure Obtained in an Experimental Evolution Process. Life, 12(2), 145. https://doi.org/10.3390/life12020145