3.1. Formation of C1 Compounds and Choosing Reference States
With the reference molecules (CO
2, H
2, H
2S and H
2O) assigned
Gr0 = 0.0 kcal/mol, we can determine
Gr0 values for CHOS species by calculating Δ
G for the formation reaction of each compound where the carbon “food” source is CO
2, the sulfur source is H
2S, additional source of hydrogen as a reductant comes from H
2, and H
2O is a byproduct of the reduction reaction. We group compounds by oxidation number of carbon, formally calculated by assigning the oxidation numbers of H (+1), O(−2) and S(−2). Thus, in CO
2, carbon has oxidation number +4. If CO
2 is fully reduced to methane (carbon in −4 oxidation state),
and therefore,
Gr0 of CH
4 is −39.5 kcal/mol. If CO
2 is not fully reduced, there are several possibilities.
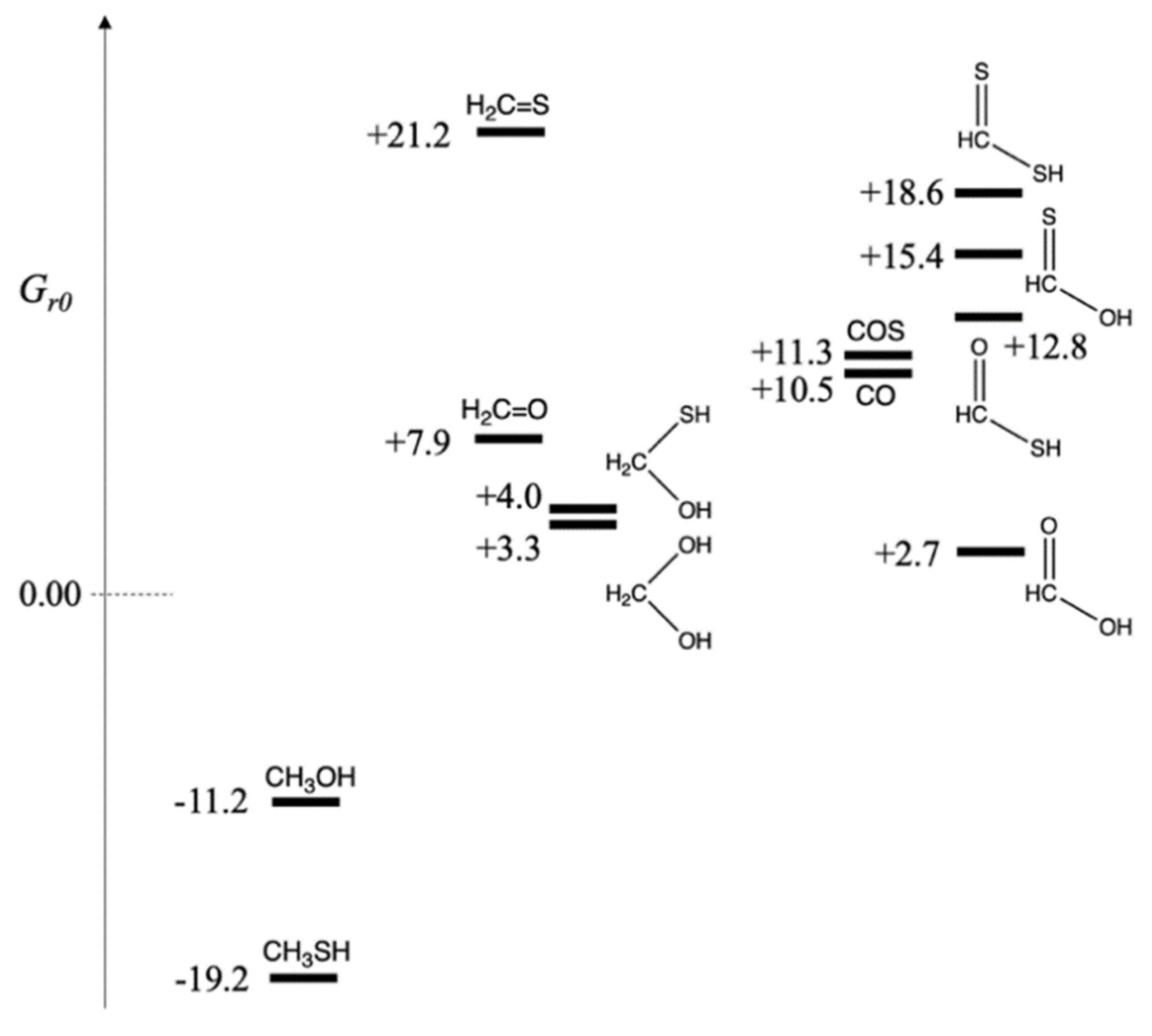
- (a)
Carbon in −2 oxidation state:
Thus, Gr0 of CH3OH and CH3SH are −11.2 and −19.2 kcal/mol respectively. Both reactions are exergonic. The thiol is more stable than the alcohol by 8 kcal/mol. In a prebiotic environment where H2S is present to reduce CO2, we expect to observe methanethiol. The relative amount of H2S versus H2 would lead to different relative ratios of CH3SH and CH3OH in the product mixture (There would of course be many other compounds!).
- (b)
Carbon in zero oxidation state:
Thus,
Gr0 of CH
2O and CH
2S are +7.9 and +21.2 kcal/mol respectively. Both reactions are endergonic, i.e., it is thermodynamically unfavorable to form formaldehyde and its thione counterpart by reducing CO
2. Forming CH
2S with its weaker C=S double bond is significantly unfavorable. However, in aqueous solution, hydration can take place across the double bond.
For the hydrates, the sulfur-containing compound is marginally less stable (by 0.7 kcal/mol) than its counterpart. Both hydrates are still slightly higher in free energy compared to the reactants, but are now likely to be accessible.
- (c)
Carbon in +2 oxidation state:
While formation of formic acid is only marginally endergonic from CO2 reduction, all three sulfur analogs are significantly higher in free energy. The thioacid, the best of the three, is 10 kcal/mol less stable than its carboxylic acid counterpart. (The two thione acids are even less stable.) This suggests that if a thioacid can be formed in some way, its hydrolysis to the carboxylic acid would be 10 kcal downhill and can be utilized to drive an uphill C–C bond-forming carboxylation reaction, typically 4–8 kcal endergonic based on our previous work. (We will see in a later section that thioesters are typically 6–7 kcal uphill from their hydrolyzed product.)
In prebiotic experiments for carbon fixation, COS has been used as an activating reagent (and the carbon source). The reaction CO2 + H2S → COS + H2O is endergonic by 10.5 kcal. Thus, we can assign Gr0 of COS as +10.5 kcal/mol. Carbon monoxide has also been used as an activated reactant in prebiotic chemistry. The reaction CO2 + H2 → CO + H2O is endergonic by 11.3 kcal. Thus, we assign Gr0 of CO as +11.3 kcal/mol. If either COS or CO are used as the carbon source rather than CO2, the formation of formaldehyde, its hydrate, or CH2(SH)(OH) are now exergonic reactions. Formic acid is also downhill ~8 kcal/mol, and the thioacid is now only marginally higher in energy (than COS or CO) and likely to be accessible.
The relative free energies of the possible C
1 compounds are shown in
Figure 1, grouped by oxidation state of carbon. On the left are the more reduced compounds, CH
3SH and CH
3OH with oxidation state of −2. In the center are CH
2O, CH
2S, and their hydrates at zero oxidation state. Furthermore, on the right are the acids with oxidation state +2. Carbon monoxide (the dehydrate of formic acid) is in this group, and because of the similar prebiotic chemistry of COS and CO, we have grouped them together. While we have formally assigned sulfur an oxidation number of −2 (so it can be grouped alongside oxygen for ease of analyzing the results), the electronegativity of sulfur is not too different from carbon. Our formal assignments are a bookkeeping method for ease of presentation, allowing us to group together compounds that only differ by swapping an S with an O or vice versa.
Figure 1 makes it clear which compounds are accessible downstream using CO or COS as the carbon source rather than CO
2. The fact that both CO and COS are ~11 kcal/mol less stable in free energy than CO
2 allows them to function as activated reactants and drive subsequent reactions along a downhill thermodynamic gradient. However, if only CO
2 was available as the carbon ‘food’ source, it is less likely that thioacids or thiones would be accessible; and the main C
1 sulfur-containing compound would be CH
3SH.
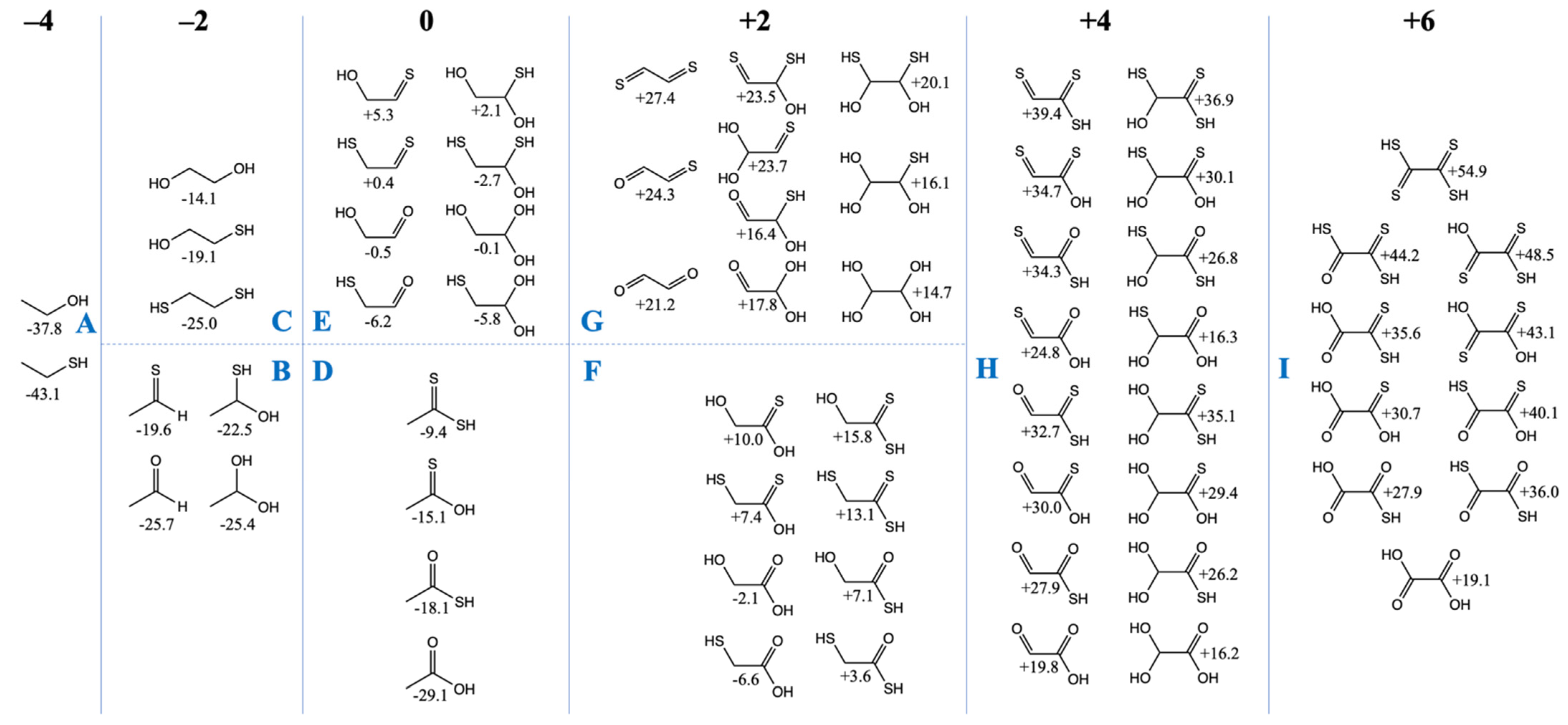
3.2. The Free Energy Landscape of C2 Compounds
We now turn our attention to the C
2 compounds of CHOS and compare them to their CHO counterparts. Do the same trends we’ve seen for the C
1 compounds hold in the C
2 cases? In
Figure 2, we have grouped the compounds according to the total formal oxidation state of the carbons, e.g., ethanethiol (CH
3CH
2SH) has six hydrogens (+1 each) and one sulfur (−2), and thus the carbons must add up to −4 for an overall neutral molecule.
Similar to our more extensive study of CHO compounds [
6],
Gr0 values are lowest for the most reduced compounds and
Gr0 values increase with oxidation. All compounds in the −4 and −2 oxidation groups have negative
Gr0 values, i.e., they are more stable relative to the reference reactants CO
2, H
2 and H
2S. Similar to the C
1 case, thiol groups are favored over alcohols. In
Figure 2A, ethanethiol is more stable than ethanol by ~5 kcal/mol, and in
Figure 2C, replacing an OH by an SH is favorable by 5–6 kcal/mol. In
Figure 2B, ethanal is 6 kcal/mol more stable than its counterpart with a C=S thione group (unlike the large gap of 13 kcal/mol in the C
1 case). For the C
2 case, hydrating the aldehyde hardly changes its
Gr0 value, while hydrating the thione stabilizes it by ~3 kcal/mol.
In the CHO compounds from our previous study [
6], having a carbonyl group was always more stable than having two separate alcohol groups (on different carbons) by a significant amount (over 10 kcal). However, this trend is reversed with sulfur; having two separate thiol groups (on different carbons) is more stable than the thione (with its weaker C=S pi-bond). For the −2 oxidation group, this leads to the most stable compounds in
Figure 2B (the aldehyde and its hydrate) having a similar
Gr0 value to the most stable compound (the dithiol) in
Figure 2C.
For the zero oxidation group, the acids (
Figure 2D) are significantly more stable than their isomers (
Figure 2E) which have separate C=X and C–XH groups. In
Figure 2D, the trend is similar to the C
1 compounds: the carboxylic acid is more stable than the thioacid by 10 kcal/mol, and the thioacid is more stable than its thione isomer by 3 kcal/mol. (The CSSH compound is further destabilized by ~6 kcal/mol.) In
Figure 2E, the most stable compound is mercaptoacetaldehyde (
Gr0 = −6.2 kcal/mol), as expected, because thiols have lower
Gr0 values than alcohols. Glycolaldehyde (
Gr0 = −0.5 kcal/mol) is close in stability to its sulfur counterpart (
Gr0 = +0.4 kcal/mol) because thiol stabilization over the alcohol is almost equally balanced by carbonyl stabilization over the thione. Hydration trends are similar to what we saw in
Figure 2B.
The two sets of compounds in the +2 oxidation group are glycolic acid with its sulfur analogs in
Figure 2F, and glyoxal with its sulfur analogs in
Figure 2G. The mercaptoacid (
Gr0 = −6.6 kcal/mol) is the most stable, followed by glycolic acid (
Gr0 = −2.1 kcal). These are the only two compounds with negative
Gr0 values in this group. Overall trends comparing the substitution of oxygen with sulfur are similar to previous cases, although we note that the gap between the thiol versus alcohol is now only 3–4 kcal/mol (instead of 5–6 kcal/mol). In
Figure 2G, the gap between a thione and aldehyde has also reduced further to ~3 kcal/mol.
In
Figure 2G (the +4 oxidation group), energy trends are similar to previous cases both for hydration reactions and for O to S substitutions in functional groups. There are two exceptions: (OH)
2CHCSSH (
Gr0 = +35.1 kcal/mol) is ~3 kcal/mol higher than expected from the general trend; and S=CC(=O)SH (
Gr0 = +34.3 kcal/mol) is ~4 kcal/mol higher than expected from the general trend. It is unclear why this is so, but we do not expect these sulfur analogs to play an important role given their very positive
Gr0 values. The most stable compounds in this group are the glyoxylic acid hydrate (
Gr0 = +16.2 kcal/mol) and its thione hydrate counterpart
Gr0 = +16.3 kcal/mol). Glyoxylic acid is an activated species in proto-metabolism, as discussed in our previous work [
6], and not surprisingly is used (as glyoxylate) experimentally to drive proto-metabolic reactions in prebiotic chemistry.
Oxalic acid (
Gr0 = +19.1 kcal/mol) is the most stable compound in the +6 oxidation group (
Figure 2I). All its sulfur counterparts have very positive
Gr0 values and they are not expected to be accessible or utilized in a sulfur-containing proto-metabolism.
3.3. Thermodynamics of C–S Coupling Reactions
Now that we have a lay of the land with our preliminary map of Gr0 values for C1 and C2 CHOS compounds, we can begin to assess the thermodynamics of forming C–S bonds if these are to play a role in proto-metabolic reactions.
In a prebiotic setting where CO
2 is reduced by a mixture of H
2 and H
2S, two CHOS C
1 compounds that we might expect to see are methanethiol (CH
3SH) and the thione-hydrate CH
2(OH)(SH). We also expect the CHO compounds methanol, formaldehyde (and its hydrate), and formic acid to be present. (Methane, the most thermodynamically favorable product, will also be present but is unlikely to react any further in a reducing environment and can be considered a “waste” molecule.) In our previous work on formaldehyde oligomerization [
11], polyols and oxanes are produced in condensation reactions forming new C–O bonds. These polyols and oxanes are marginally unfavorable thermodynamically compared to the monomer (hydrate) but the free energy difference is very small. How does forming new C–S bonds fare?
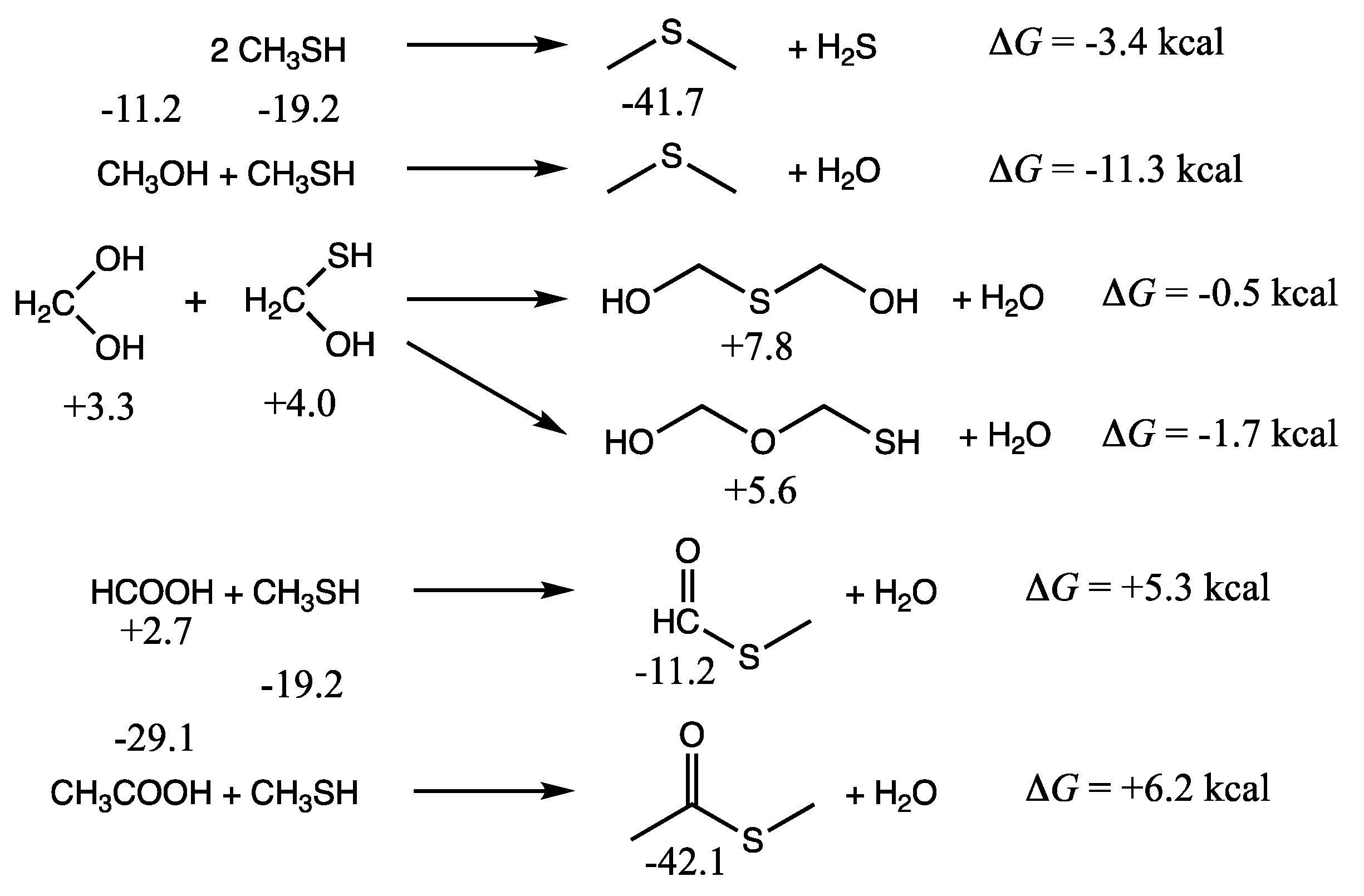
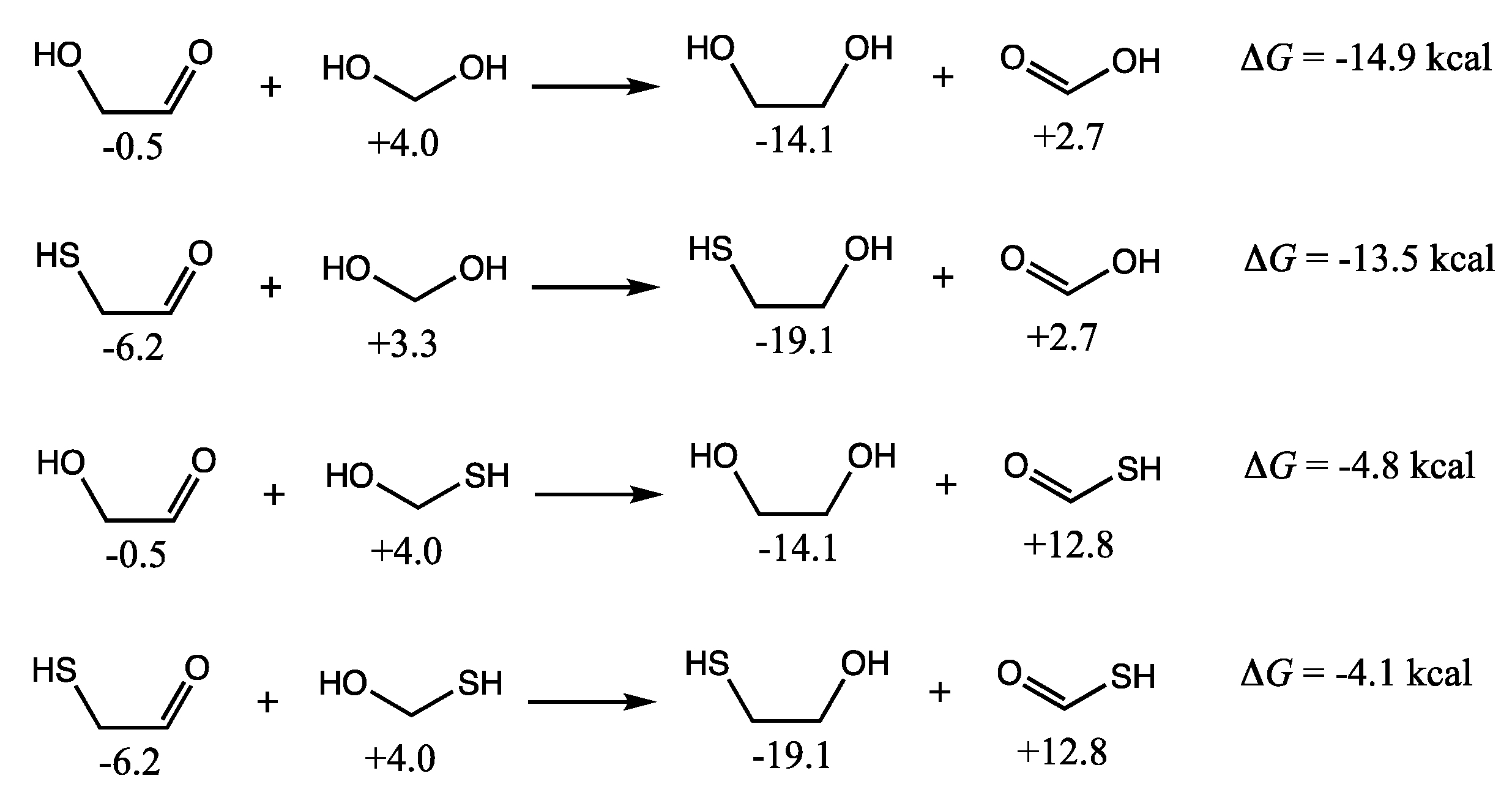
As shown in the first two reactions of
Figure 3, the formation of dimethylsulfide from methanethiol is exergonic. We calculate Δ
G of the reaction by subtracting
Gr0 of the reactants from
Gr0 of the products. (Recall that reference molecules have zero
Gr0 values.) Thus, Δ
G = (−41.7 + 0.0) − 2(−19.2) = −3.4 kcal. The condensation of methanethiol and methanol to form dimethylsulfide is more exergonic: Δ
G = (−41.7 + 0.0) − (−11.2 + (−19.2)) = −11.3 kcal. It is certainly more favorable than forming dimethylether (Δ
G = +5.5 kcal). Thus, we expect dialkylsulfides to be formed if methylsulfide is present.
If hydrated formaldehyde and its counterpart C
1 thione are present, their condensation reactions are mildly exergonic, and so we might expect to see HO–CH
2–X–CH
2–XH compounds (X = O or S) as shown in the middle set of reactions in
Figure 3. Forming the C–O–C compound is marginally more favorable than the C–S–C in this case. Thus, one might expect to see mixed polyol/polythiols depending on the concentrations of monomers. In an aqueous solution, the equilibrium will shift towards hydrolysis back into the monomers. For a 1 M solution, where water molecules outnumber solutes by 55:1, the correction factor is 2.4 kcal/mol in favor of hydrolysis [
13]. We expect that for dilute solutions, monomers will be favored over condensation reactions that form C–X–C bonds (while releasing water), and hence we have not pursued calculating the free energies of polythiols or thiolanes. For the energetics of oxane/polyol formation from formaldehyde, the reader can refer to our previous work [
11].
The final pair of reactions in
Figure 3 illustrate thioester formation from the reaction of CH
3SH with formic or acetic acid. These reactions are endergonic by 5.3 and 6.2 kcal respectively. In contrast, as we saw in the previous two sections, thioacid formation is endergonic by ~10 kcal. Since compounds with thiol groups are thermodynamically favored over their alcohol counterparts, and carboxylic acid groups (if they can be formed) are the most stable compounds in an oxidation group, this hints towards the role of thioesters in a prebiotic milieu as an important intermediate in chemical processes that couple endergonic and exergonic reactions.
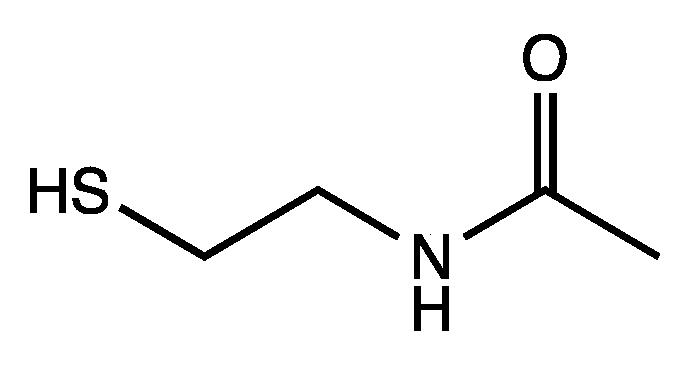
In extant biochemical reactions involving the coenzyme CoA, forming the thioester is typically ~7 kcal uphill. Using the small molecule analog shown in
Figure 4, we calculate that its condensation with acetic acid and succinic acid are +6.9 kcal and +7.5 kcal respectively. Thus, exergonic hydrolysis of such thioesters can potentially be coupled to proto-metabolite C–C bond formation where the carboxylation reactions are endergonic by 4–7 kcal, as shown in our previous work on CHO systems [
6]. Our preliminary results, while promising, would not do justice to the complexity of the system, and we expect to provide a detailed examination of the connection between thioesters and potential CHO proto-metabolic systems in a future publication.
3.4. Sulfur Analogs of the Formose Reaction
In extant biochemical cycles, the reduction of CO
2 to build biomass can proceed through cycles analogous to the reverse TCA cycle. We explored the thermodynamics of four such cycles in CHO systems in our previous work [
6], the most interesting being the 3-hydroxypropionate/4-hydroxybutyrate (3HP/4HB) cycle because it does not involve CHO compounds with more than four carbons and avoids forming the less stable oxaloacetate. In that work, we proposed alternative pathways that could be thermodynamically more feasible than the 3HP/4HB cycle thereby avoiding some of the more challenging kinetic barriers, but we also noted that in the absence of (specialized) enzyme catalysts there would still be kinetically unfeasible steps in a prebiotic milieu.
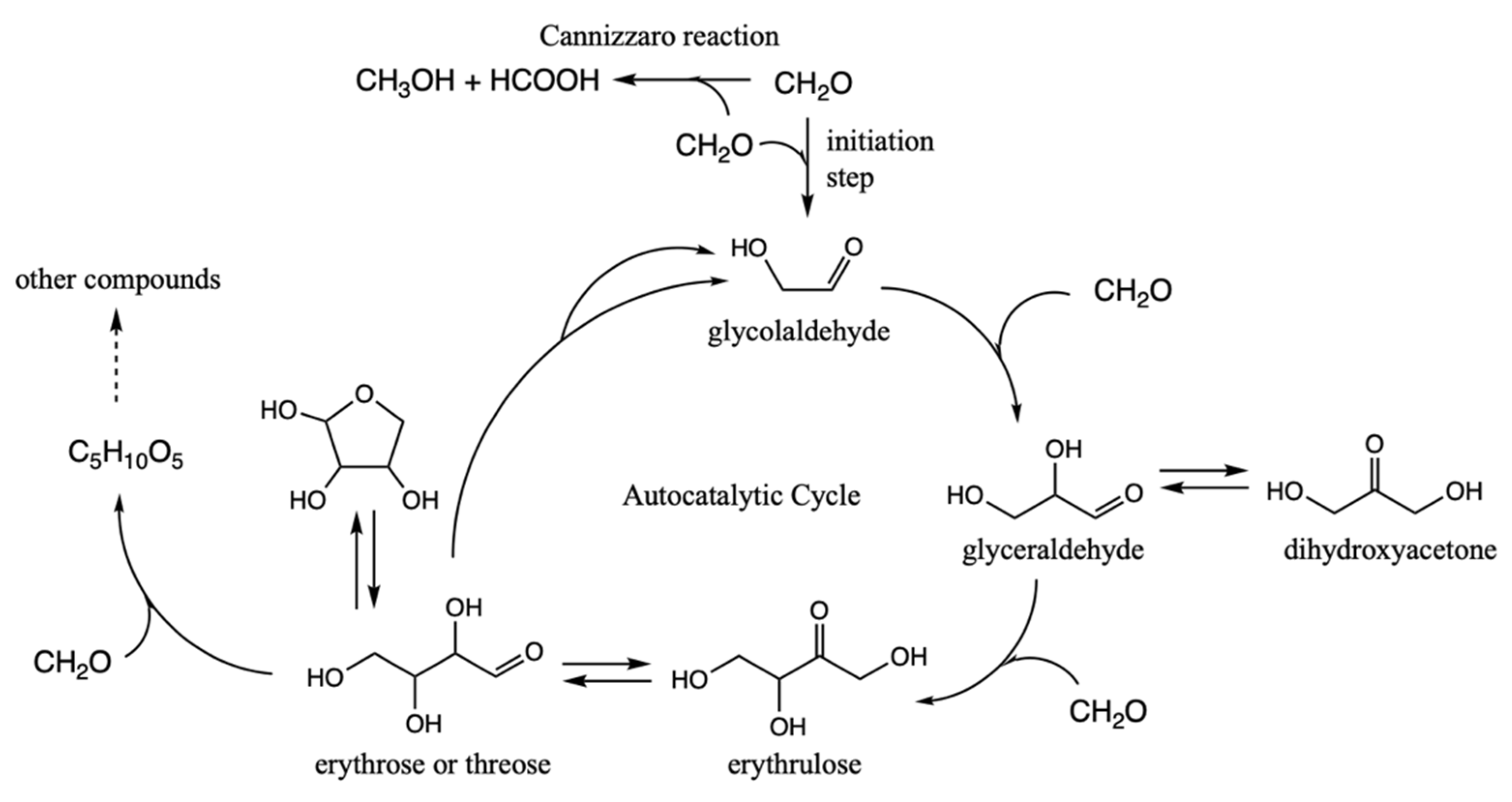
There is a known autocatalytic reaction that builds up progressively larger CHO compounds from a C
1 species—the formose reaction [
7]. It takes advantage of aldol reactions to form new C–C bonds, and autocatalysis is aided by a retro-aldol transformation of a C
4 species into two C
2 compounds. It is thus analogous to the 3HP/4HB cycle, but much simpler because it avoids redox reactions: formaldehyde is the C
1 ‘food’ species, glycolaldehyde is the linchpin C
2 species, and all compounds involved remain in the zero oxidation group. In contrast for the 3HP/4HB cycle, while acetate (the C
2 linchpin) is in the zero oxidation group, CO
2 (+4 oxidation group) is the C
1 food species and therefore reducing equivalents of H
2 are required for the cycle to be realized.
The problem with the formose reaction is that it is a mess [
8], and a slew of compounds are formed in an essentially uncontrolled reaction. Could the presence of sulfur introduce some form of thermodynamic control to the reaction? How might the kinetics change? Is there a path towards taming the formose reaction as a stepping stone towards proto-metabolic cycles that more closely resemble what extant life uses? Building on what we have learned from our survey of CHOS C
1 and C
2 compounds described earlier, this subsection presents our free energy map of sulfur analogs to the formose reaction. A brief summary of the key compounds in the (non-sulfur-containing) formose reaction are shown in
Figure 5.
In discussing the results, we will repeatedly make reference to our earlier free energy map of the thermodynamics and kinetics of the formose reaction (up to C
4); this paragraph provides the highlights from that work [
11]. Forming glycoaldehyde directly from CH
2O is very challenging kinetically. We previously calculated the barrier for direct dimerization to be 45.3 kcal. Experimentally, in a solution only containing CH
2O, there is a long induction period. However once even a small amount of glycolaldehyde is formed (or added to the solution as an initiator), the reaction proceeds rapidly producing a wide variety of sugars, mostly in the C
4 to C
7 range. With C
2 present, the difficult C
1 + C
1 → C
2 reaction is bypassed by the much lower barrier C
2 + C
1 → C
3 and C
3 + C
1 → C
4 reactions. The retro-aldol C
4 → C
2 + C
2 reaction regenerates (more) C
2 and accelerates the consumption of C
1 making the cycle autocatalytic. CH
2O can also form polyols and oxanes but hydrolysis in an aqueous solution favors re-forming the monomer. On the other hand, the Cannizzaro disproportionation reaction parasitizes the cycle (to be discussed in a later subsection of this paper). Extensive documentation of experimental results on the formose reaction can be found in a long article by Mizuno and Weiss [
27].
If H
2S was present as a source of sulfur, one might expect a starting mixture of the hydrates CH
2(OH)
2 and CH
2(SH)(OH) in aqueous solution, as they are relatively close in energy with
Gr0 values of +3.3 and +4.0 kcal respectively. Our calculated barrier for the direct C–C coupling reaction of CH
2O and CH
2S is 26.0 kcal, which is much lower than 45.3 kcal for the direct dimerization of CH
2O, but recall from
Figure 1 that CH
2S is 13.3 kcal/mol less stable than CH
2O, which accounts for two-thirds of the difference. Mercaptoacetaldehyde (
Gr0 = −6.2 kcal) is the C
2 species formed, and the reaction is thermodynamically favorable (Δ
G = −19.7 kcal from the hydrates). Since a range of C
1 and larger species (C
2, C
3, etc.) are observed experimentally in prebiotic reactions [
28,
29,
30,
31,
32,
33] by reducing CO
2 (or bicarbonate or CO or COS) simulating hydrothermal vent prebiotic chemistry, and since the C
1 + C
1 → C
2 initiation step is not important for the cycle, we need not worry about the initiation step. Our starting point will be the C
2 linchpin species, mercaptoacetaldehyde, the thiol analog of glycolaldehyde. Mercaptoacetaldehyde has also been proposed as central in prebiotic scenarios involving the amino acid cysteine [
34].
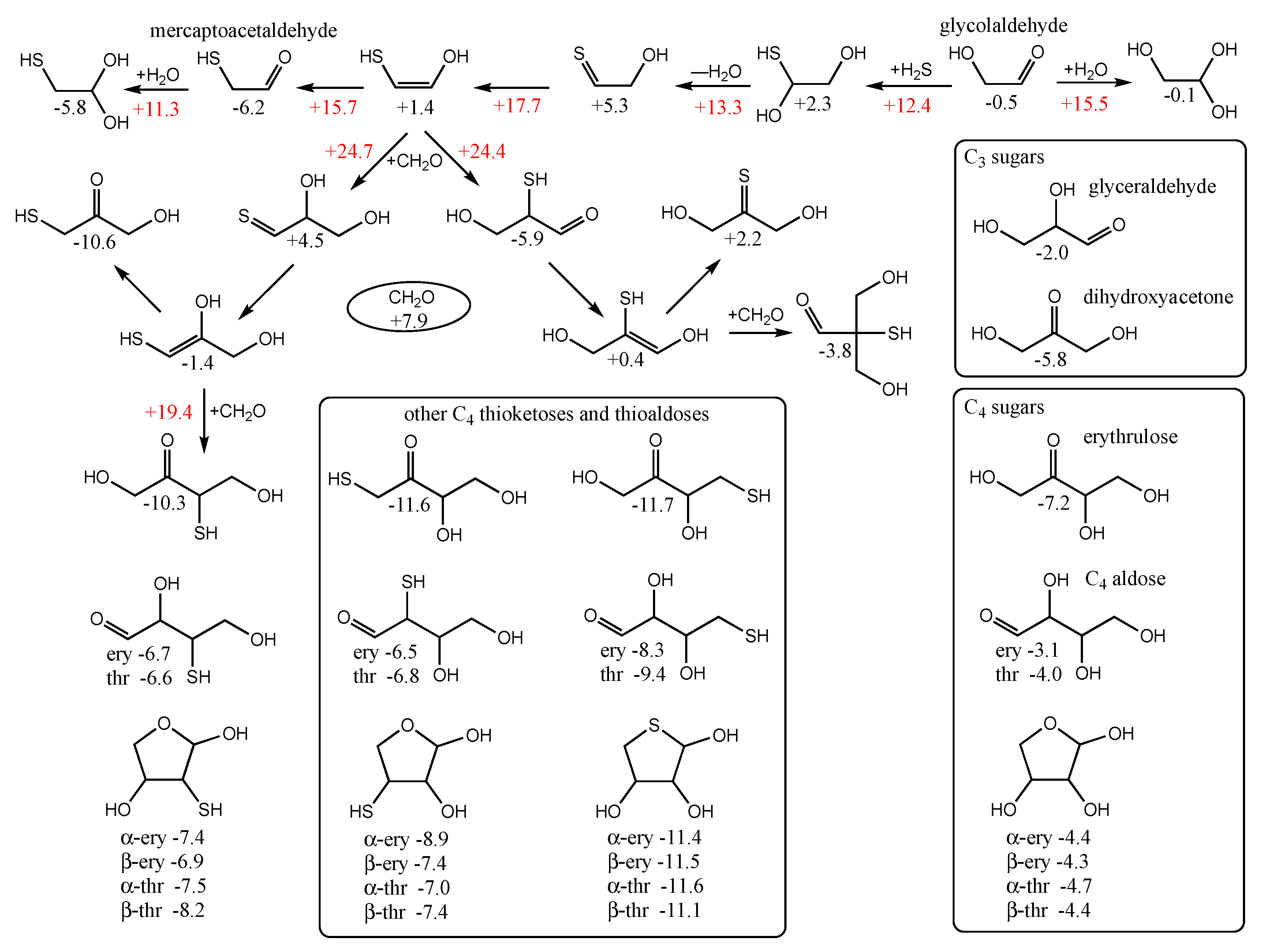
Since
Gr0 = −6.2 kcal/mol for mercaptoacetaldehyde, it is favorable thermodynamically to be (one among many possible compounds) produced prebiotically from a source containing CO
2, H
2 and H
2S. (It may not be as easily observed experimentally because it participates in further reactions.) Mercaptoacetaldehyde can also potentially be formed from glycolaldehyde in the presence of H
2S as shown in the top row of
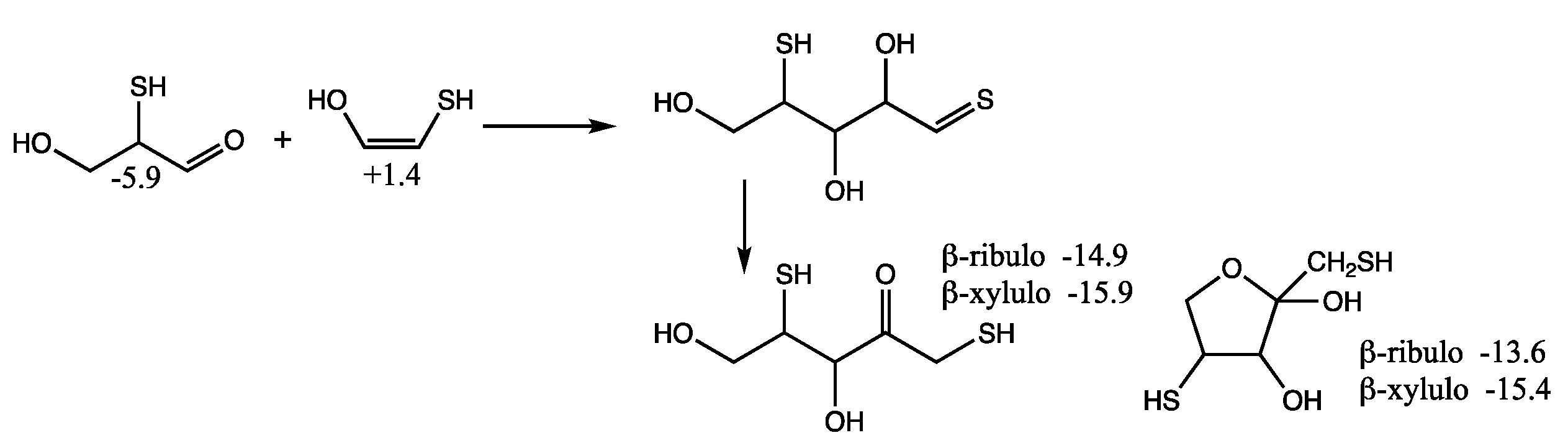
Figure 6. The reaction is overall thermodynamically favorable, Δ
G = −6.2 − (−0.5) = −5.7 kcal. Note that the cis-enol of mercaptoacetaldehyde as shown in
Figure 6 is more stable than the trans-enol (not shown) by ~2 kcal/mol in our calculation of
Gr0.
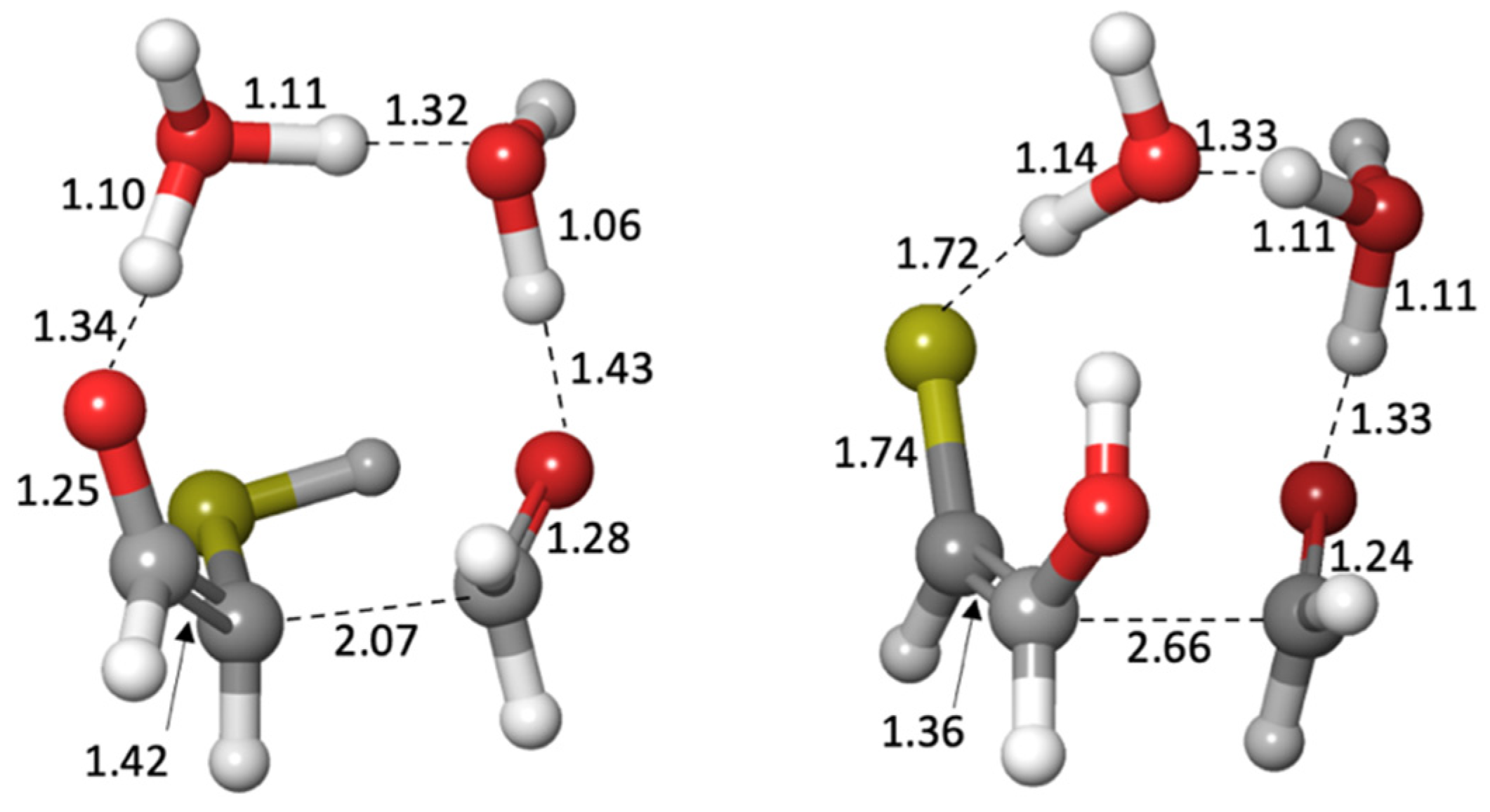
By calculating the energies of the transition states (
Gr0 values in red next to arrows), we can estimate the reaction kinetics. For example, the first step of adding H
2S to glycolaldehyde has a barrier of +12.4 − (−0.5) = 12.9 kcal. (The corresponding dehydration barrier in the reverse reaction is +12.4 − (+2.3) = 10.1 kcal.) The calculated stepwise barriers for the overall transformation of glycolaldehyde to mercaptoacetaldehyde (involving formation of the thione intermediate and its enol) are in the 11–14 kcal range. (At both ends on the top row, we also show the hydration reactions of mercaptoacetaldehyde and glycolaldehyde for completeness; see
Supplementary Materials for transition state structures.)
In our previous work on CH
2O oligomerization [
11], aldol additions of CH
2O proceed via the enol. We see the same for mercaptoacetaldehyde, except that its asymmetry allows for two possible products: the less favorable thione (
Gr0 = +4.5 kcal) and the more favorable aldehyde (
Gr0 = −5.9 kcal) that has a thiol on the central carbon. Kinetically, we might also expect the aldehyde to be favored because the thiol carbon of the enol is a better nucleophile than the alcohol carbon. However, our calculated barriers are essentially identical; this is after optimizing multiple transition states and the lowest energy structures are shown in
Figure 7.
Considering the enol (Gr0 = +1.4 kcal) and CH2O (Gr0 = +7.9 kcal) as the reactants, the barrier to forming the C3 aldehyde is 24.4 − (1.4 + 7.9) = 13.3 kcal, and the barrier to the C3 thione is 24.7 − (1.4 + 7.9) = 13.6 kcal. If mercaptoacetaldehyde and CH2O were the reactants, the barriers would, respectively, be 24.4 − (−6.2 + 7.9) = 22.7 kcal and 24.7 − (−6.2 + 7.9) = 23.0 kcal. These calculated barriers are very similar to our previous work for the C1 + C2 → C3 aldol addition of glycolaldehyde and CH2O of 22.3 kcal (or 13.0 kcal from the enol). Thus, in a mixture that contained glycolaldehyde, mercaptoacetaldehyde, and CH2O, the kinetics for this first aldol addition (C1 + C2 → C3) are similar and both C2 “reactants” will consume the C1 food source (CH2O) at similar rates.
Let us now consider the thermodynamics of the C1 + C2 aldol addition. In the CHO system, forming glyceraldehyde (Gr0 = −2.0 kcal) is favorable with ΔG = −2.0 −(−0.5 + 7.9) = −9.4 kcal. The analogous reaction in the CHOS system with mercaptoacetaldehyde, forming the C3 aldehyde-thiol, is similarly favorable: ΔG = −5.9 − (−6.2 + 7.9) = −7.6 kcal. On the other hand, forming the thione-diol is slightly unfavorable: ΔG = +4.5 − (−6.2 + 7.9) = +2.8 kcal. Since the C3 aldehyde-thiol has the lower Gr0 value, it could be thermodynamically favored over glyceraldehyde in an equilibrating mixture with multiple reactants.
However, the situation is more complicated because “globally” among the C
3 structures, the thioketose (
Gr0 = −10.6 kcal, leftmost structure in the second row of
Figure 6) is the most stable, and access to it via enolization comes from the less thermodynamically favorable aldol addition. The intermediate enol with a terminal thiol (
Gr0 = −1.4 kcal, leftmost structure in the third row of
Figure 6) is also the starting point for further aldol addition of CH
2O to form the linear C
4 thiosugars. On the other hand, the enol of the C
3 aldehyde-thiol would lead to a branched C
4 thiosugar (
Gr0 = −3.8 kcal), assuming our earlier argument that the thiol carbon of the enol is the better nucleophile. However, as we saw for C
1 + C
2 → C
3, addition to the alcohol side of the enol is just as viable kinetically, and likely more so in this case to avoid steric hindrance. Thus, access to the linear C
4 thio-sugars is possible through both branches. What role might the C
3 thioketose play? Analogous to dihydroxyacetone, as discussed in our previous work [
6], it may be an “off-cycle” compound that forms an equilibrating pool of inter-connected compounds [
35] that could stabilize the cycle and provide a form (albeit simple) of regulatory control. (Dehydrations of C
3 sugars may also be a part of this pool; see
Supporting Materials).
The C
1 + C
3 addition to form the C
4 thioketose (
Gr0 = −10.3 kcal/mol, left side of
Figure 6) is thermodynamically favorable with Δ
G = −10.3 − (−10.6 + 7.9) = −7.4 kcal, very similar to the aforementioned C
1 + C
2 → C
3 addition of Δ
G = −7.6 kcal. The barrier for the C
1 + C
3 → C
4 aldol addition is 19.4 − (1.4 + 7.9) = 8.1 kcal from the enol, noticeably lower than 13.3 kcal in the analogous C
1 + C
2 → C
3. In our previous work on the CHO system [
11] (leading to erythrulose), the barrier is 8.5 kcal for C
1 + C
3 → C
4, which is similarly lower than the 13.0 kcal barrier for C
1 + C
2 → C
3. Thus, kinetically, we expect the CHOS analog of the formose reaction to show similar behavior as the CHO system under appropriate experimental conditions that facilitate the reaction. Thermodynamically (left side of
Figure 6), the thioketose is ~4 kcal more stable than its open-chain thioaldoses, while the ring structures are ~1 kcal more stable than the open thioaldoses. Once again, this is similar to the non-sulfur analogs (bottom right box in
Figure 6) of erythrulose, erythrose, threose, and the ring structures. We can think of the ketose, the open chain aldose, and the furanose as an equilibrating pool of compounds.
For the C
4 sugars, the 3-thioketose turns out to be marginally less stable than both the 1-thioketose and 4-thioketose that have terminal thiols (
Figure 6, central lower box). As for the aldoses, the 3-thioaldose and 2-thioaldose have similar energies, while the 4-thioaldose with its terminal thiol is the most stable. This is also true for the ring structures, and interestingly the 4-thioaldose rings (
Gr0 values of −11.1 to −11.6 kcal/mol) are similar in stability to the 4-thioketose (
Gr0 = −11.7 kcal/mol). This suggests that a possible role played by (terminal) thiol groups in a prebiotic setting is to stabilize the corresponding aldose rings.
A key autocatalytic step in the formose reaction is the retroaldol reaction of the C4 aldose back into two C2 linchpin molecules. In the CHO system, this reaction starting from threose is marginally uphill with ΔG = 2(−0.5) − (−4.0) = +3.0 kcal. In the sulfur analog, the thioaldose splits into mercaptoacetaldehyde and glycolaldehyde. For 4-thiothreose, we see a similar result: ΔG = (−0.5) + (−6.2) − (−9.4) = +2.7 kcal. However, for 3-thiothreose, the reaction is now energetically neutral with ΔG = (−0.5) + (−6.2) − (−6.6) = −0.1 kcal. (2-thiothreose shows a similar result with ΔG = +0.1 kcal). Thus, considering only the C4 species for the moment, we might expect over time a depletion of the 2- and 3-thio-sugars, and possible accumulation of the 4-thiosugars, favoring the aldose rings that are more resistant to hydrolysis. The reality would be a lot messier with other aldol and retro-aldol reactions occurring, alongside Cannizzaro side-reactions.
Stepping back to look at the overall thermodynamic map, we see that the C3 and C4 species show similar trends as the C1 and C2 species discussed earlier. Compared to the reference compounds, thiol groups are favored over their alcohol counterparts and are most stable in the terminal position. Thiones with their weaker C=S bonds are less stable than their carbonyl counterparts. We also have preliminary data (for a future publication) showing that the trends for sulfur analogs for the larger molecular acids mirror those for we previously discussed for the smaller molecules. Overall, we see many similarities for both the thermodynamics and kinetics when comparing individual steps in the formose reaction of the CHO system to its sulfur analogs.
3.5. Can Dithiol Groups Influence Sugar Formation?
Could having a thiol group in a sugar make a relevant and interesting difference? One possibility we explore in this subsection is based on the experimental work of Eschenmoser and colleagues [
36], where they found that starting with glycolaldehyde-2-phosphate and formaldehyde led to a higher yield of ribose among the pentose-2,4-diphosphates formed. If phosphate can “direct” the reaction to favor certain products over others (in a messy formose-like reaction), can sulfur do the same? If sulfur was primordial to phosphate in prebiotic systems, could it have played an analogous role?
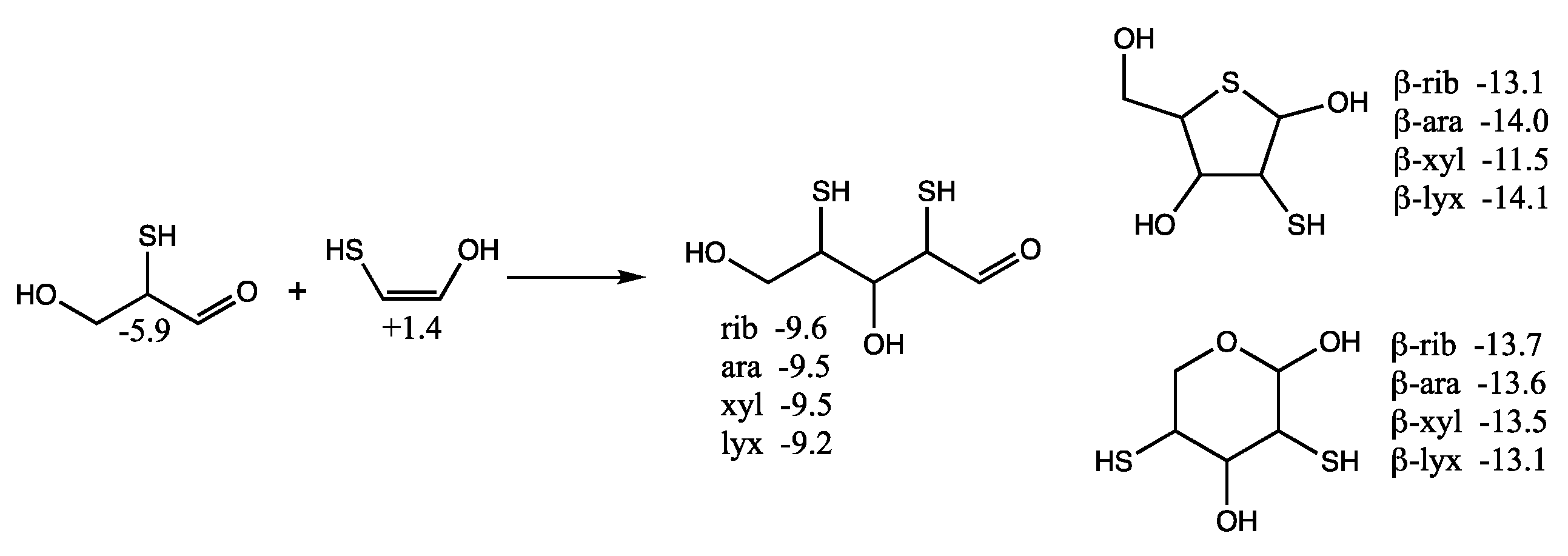
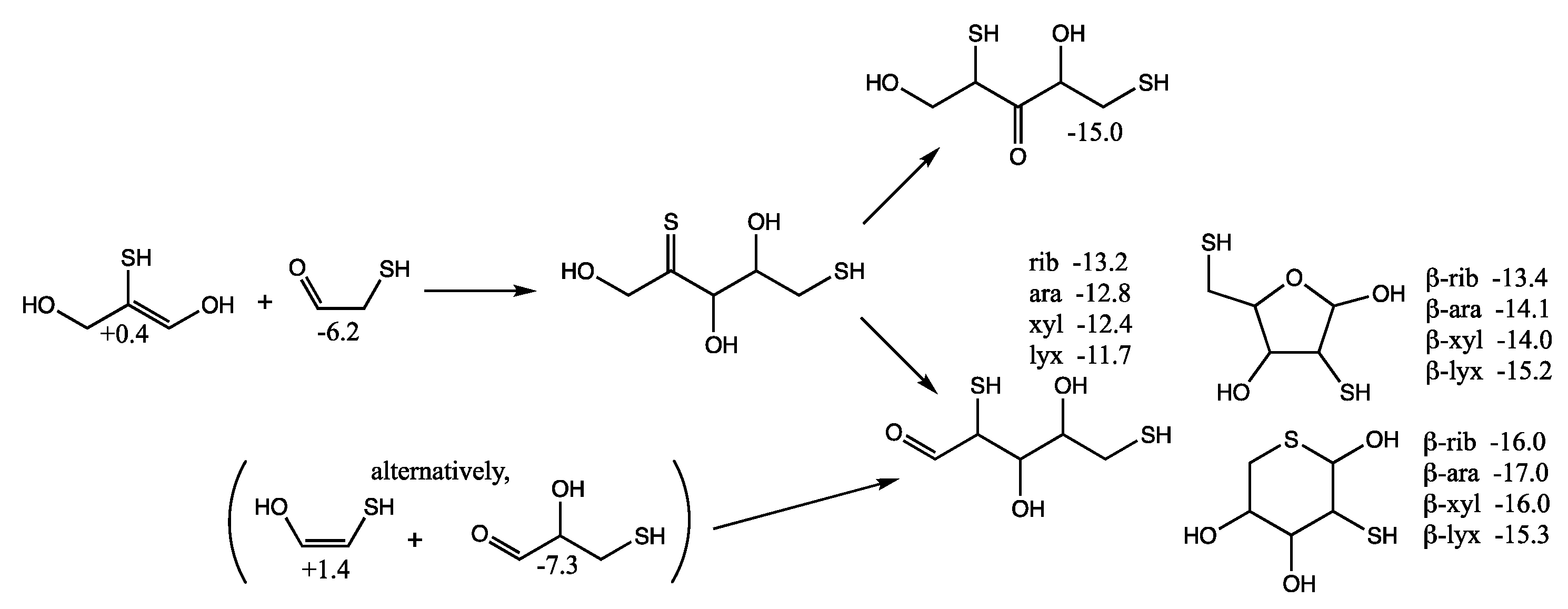
Considering mercaptoacetaldehyde as the sulfur analog of glycolaldehyde-2-phosphate, in the presence of formaldehyde we expect aldol addition to favorably form the C
3 aldehyde-thiol (as discussed in the previous section), i.e., the analog of glyceraldehyde-2-phosphate. Aldol addition of mercaptoacetaldehyde (via its enol) with the C
3 aldehyde-thiol leads to 2,4-dithioaldoses (the sulfur analogs of the C
5 aldose-2,4-diphosphates) as shown in
Figure 8. The rings are more stable than the open chain structures. Unlike the CHO sugars, the pyranoses are not more stable than the furanoses but have comparable free energies. This is because having sulfur in the ring provides a 2–3 kcal/mol stabilization (as seen for the thiotetroses in
Figure 6).
For the open chain pentoses, our calculated Gr0 values have ribose being the most stable followed by arabinose, xylose, lyxose. However, the difference in free energy is tiny and certainly within the computational error; we cannot claim that incorporation of sulfur favors ribose over the other aldopentoses. For the β-pyranoses, we see the same order of stability as the open chain structures, and again the differences are tiny and within the computational error. For the β-furanoses, arabinose and lyxose have lower Gr0 values than ribose with xylose being the least stable. We have no explanation why this is or if this is some artifact of the calculation (possibly not finding the best conformers in some cases).
Although we expect the C
2 + C
3 addition to form the C
5 2,4-dithioaldoses to be kinetically favored (because the thiol carbon of the enol is more nucleophilic than the alcohol carbon), we also consider the possibility of forming 1,4-dithio-2-ketopentose since it leads to more thermodynamically favored ketoses as shown in
Figure 9. (We have switched the position of the OH and SH in the enol in
Figure 9 compared to
Figure 8 to make the aldol addition clearer.) The thione intermediate formed is likely to isomerize to the more favored keto form. Our calculated
Gr0 values have the open chain sulfur analog of xylulose more stable than the ribulose by 1 kcal/mol. The furanoses are not as stable as the open chain structures.
However, there is another possibility. If C
3 acts as the enol (rather than the C
2), a C
5 thione intermediate can be formed that could subsequently isomerize into a 1,4-dithio-3-ketose or a 2,5-dithioaldose, as shown in
Figure 10. The most sTable 3-ketose has
Gr0 = −15.0 kcal/mol (its diastereomer is only 0.3 kcal less stable). For the open chain 2,5-dithioaldoses, the ribose analog is the most stable followed by arabinose, xylose, and lyxose. The furanoses, with a pendant thiol in the 5-position are slightly more stable than the open chain structures. The pyranoses with sulfur in the ring are the most stable group. For the ring structures, we have no explanation for the relative ordering of the most stable different stereoisomers according to our calculated
Gr0 values.
An alternative route (see parenthesis in
Figure 10) to the C
5 2,5-dithioaldoses is the aldol addition of the C
2 enol with 3-thioglyceraldehyde (
Gr0 = −7.3 kcal/mol), the isomer of the C
3 ketone (the most stable C
3 sulfur analog in
Figure 6 with
Gr0 = −10.6 kcal/mol). If thiols could be precursors to phosphates in a prebiotic world, the 2,5-dithioribose analog could be a stand-in for ribose-2,5-diphosphate.
There are other possible products from aldol additions of these sulfur analogs that we have not discussed. (See
Supplementary Materials). For example, in the C
2 + C
3 → C
5 addition, one of the species may not contain sulfur, and this would lead to a range of sugars with just one thiol group rather than two. As a second example, we have not discussed the C
4 + C
1 → C
5 addition, which would lead to other isomers such as 3,5-dithio-2-ketoses, 1,3-dithio-2-ketoses and 3,5-dithioaldoses. Furthermore, we have mainly focused on non-branched sugars, and we have only shown one example, the branched tetrose in
Figure 6. We expect these thermodynamically less stable branched sugars to be less prevalent than their straight-chain counterparts.
Our limited foray into sulfur analogs of the formose reaction is clearly not exhaustive. Our goal here is to provide a flavor of the myriad possible reactions, intermediates, and products, in this system. Based on our limited analyses, we can draw some general conclusions. Substituting an alcohol with a thiol group is thermodynamically favorable. Thiol groups in the terminal position are particularly favored. Sulfur in the sugar ring is thermodynamically favored. Furthermore, the presence of sulfur provides some asymmetry to the aldol addition reactions, and the lower electronegativity of sulfur means that in an enol, the thiol carbon is a better nucleophile which may provide some “directing” ability that favors some subsets of products over others.
While we have speculated about the possibility that thiols might be precursors to phosphates in aldol reactions of sugars, our results thus far are inconclusive on this topic. However, there are tantalizing analogies. In the pentose phosphate pathway, the sugars involved have terminal phosphates, and our limited study finds that terminal thiols are thermodynamically favored. By including thiols in the mix, we find that aldoses can be as thermodynamically stable as ketoses for C4 and C5, while this is not so in CHO sugars where the ketose is typically 2–3 kcal more stable than the aldose.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










