
Utilization of Genotyping-by-Sequencing (GBS) for Rice Pre-Breeding and Improvement: A Review

 , , and
, , and
Abstract
1. Introduction
2. Advances in Chemistry of GBS
2.1. Restriction Enzyme-Based (RE-Based) GBS
2.2. PCR-Based GBS
2.3. Target Capture
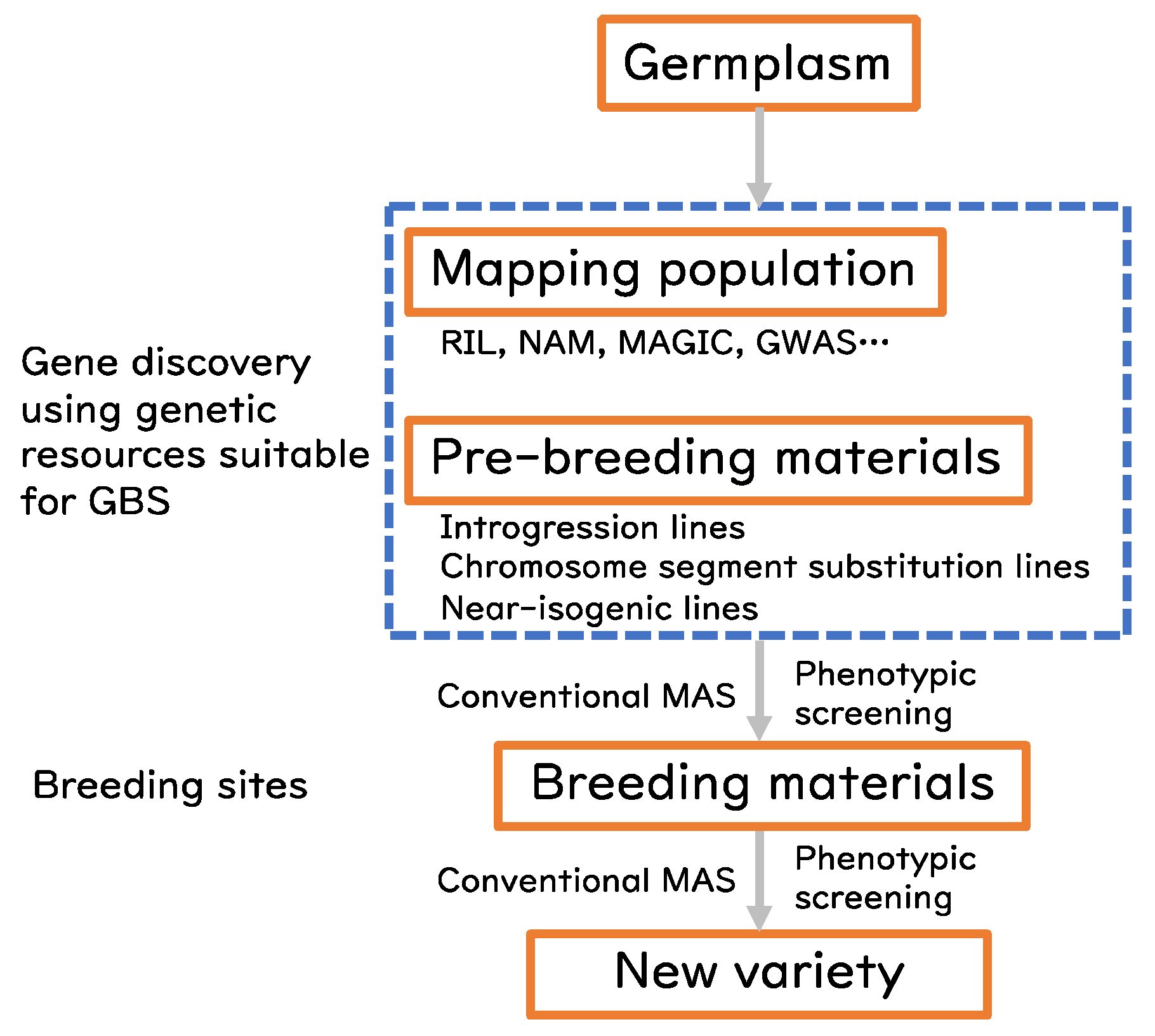
3. GBS for Rice Pre-Breeding
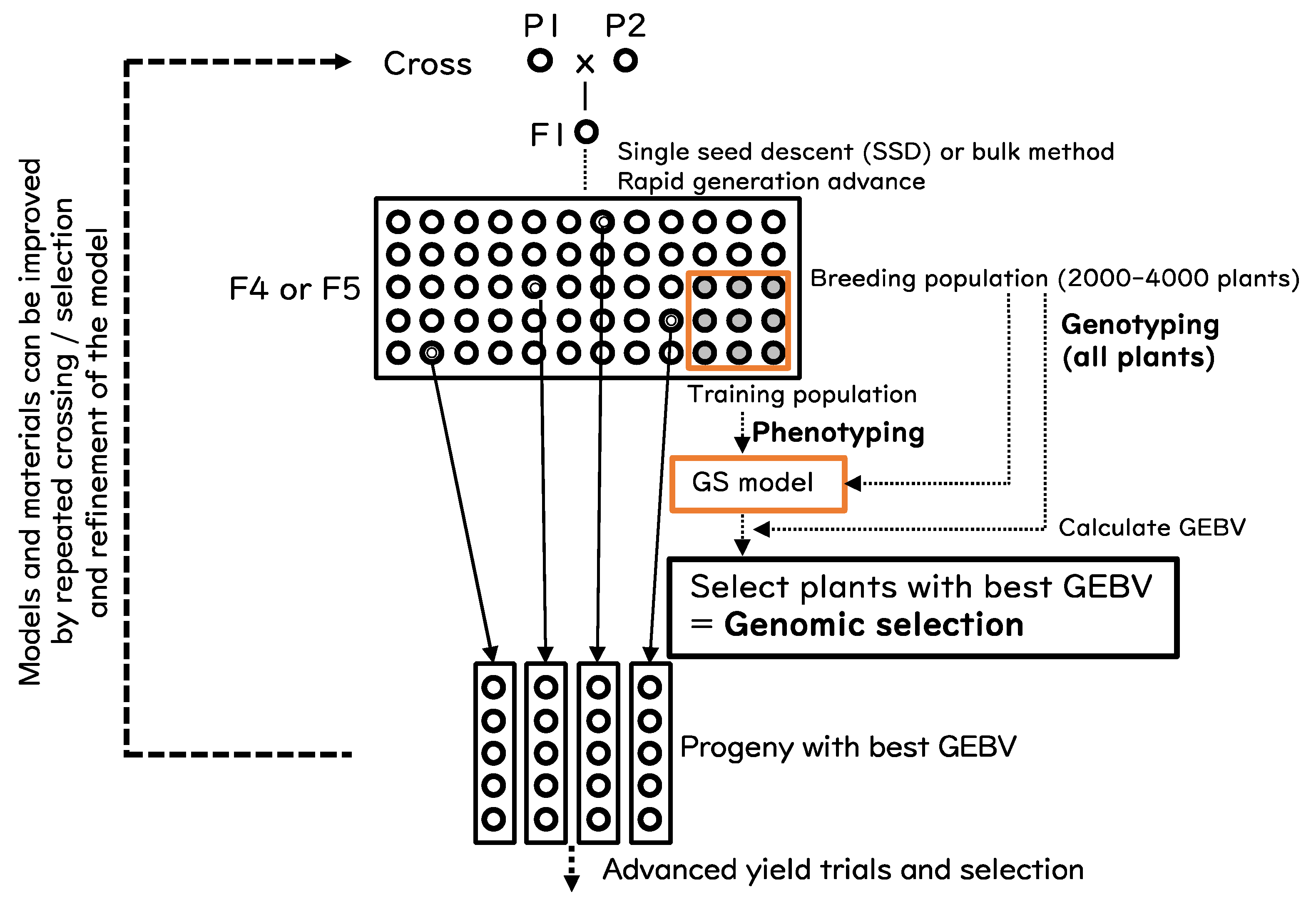
4. Implementation of GS and GBS for Rice Improvement
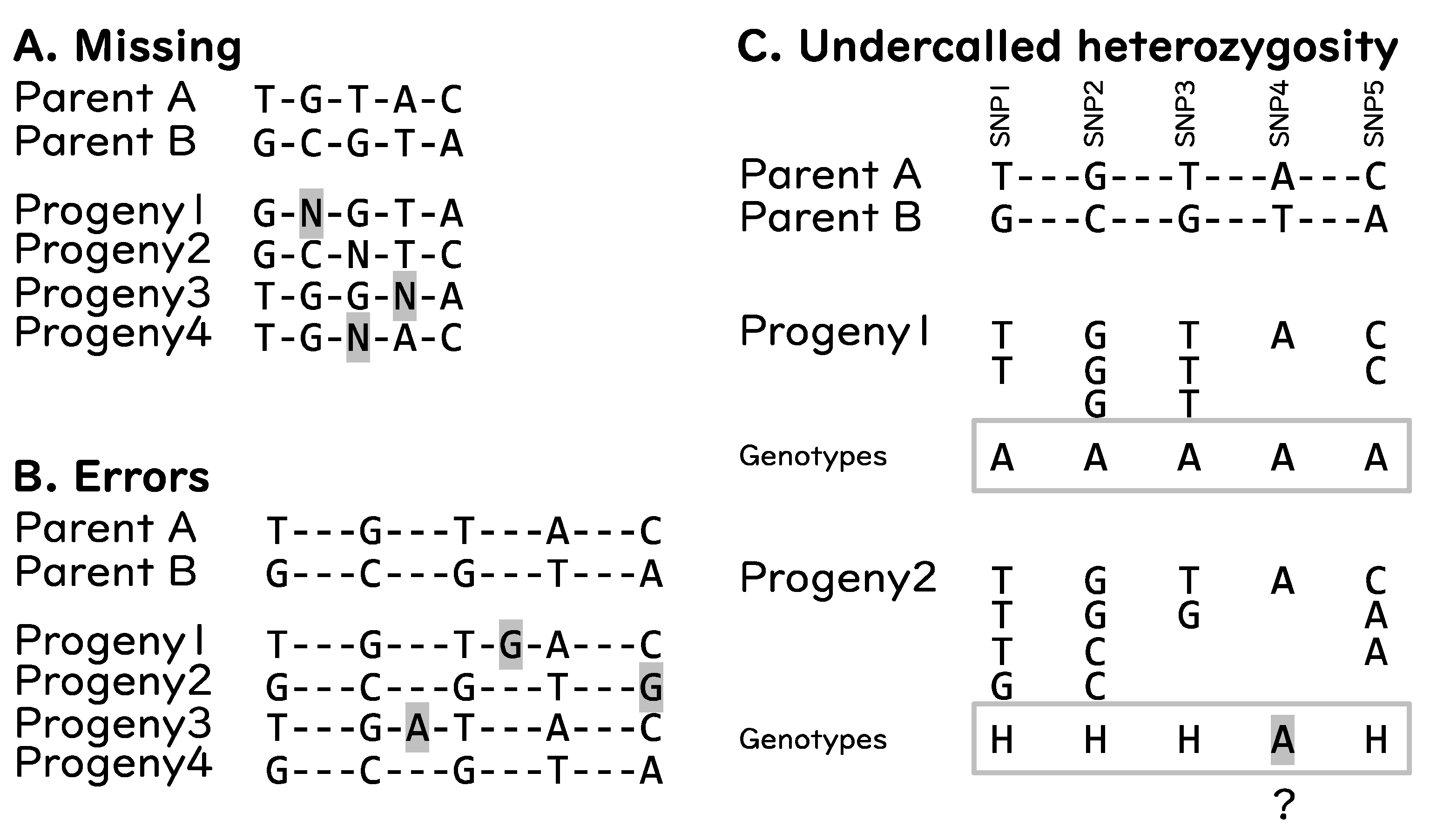
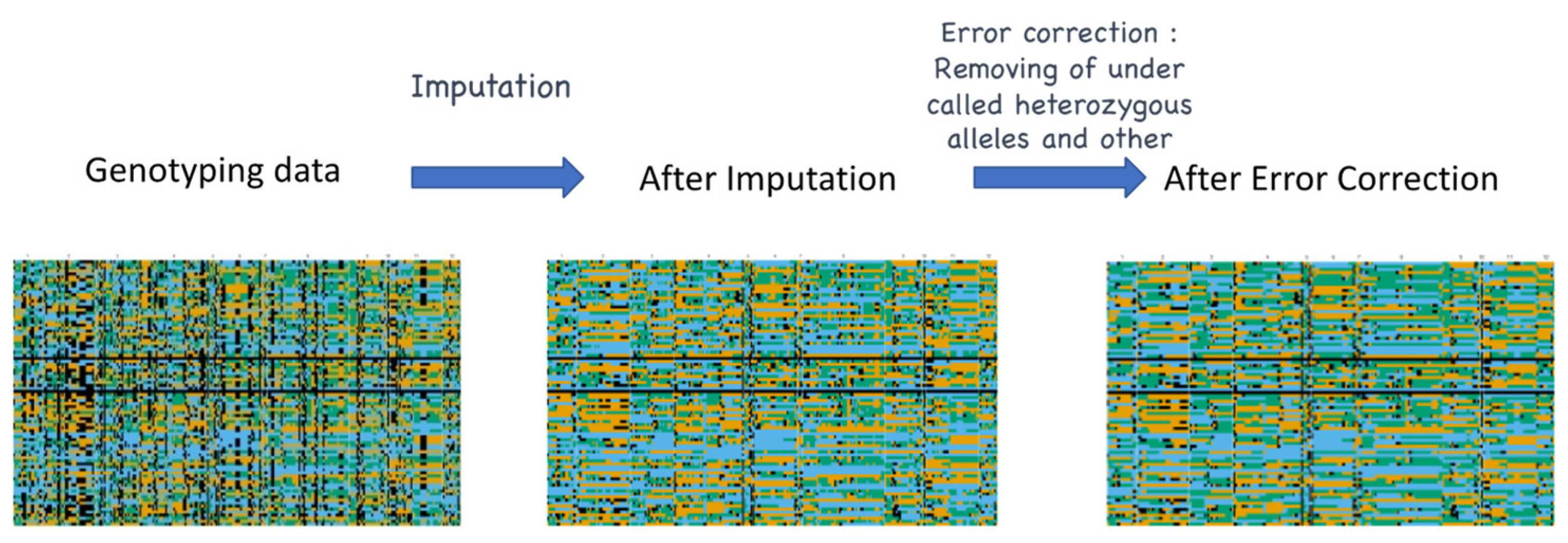
5. Challenges in Informatics
6. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Rice Genome Sequencing Project; Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ashikari, M.; Matsuoka, M. Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends Plant Sci. 2006, 11, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a Major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef]
- Doi, K.; Izawa, T.; Fuse, T.; Yamanouchi, U.; Kubo, T.; Shimatani, Z.; Yano, M.; Yoshimura, A. Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev. 2004, 18, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Brar, D.S.; Dalmacio, R.; Elloran, R.; Aggarwal, R.; Angeles, R.; Khush, G.S. Gene transfer and molecular characterization of introgression from wild Oryza species into rice. In Rice Genetics III (In 2 Parts), Proceedings of the Third International Rice Genetics Symposium, Manila, Philippines, 16–20 October 1995; Khush, G.S., Hettel, G., Rola, T., Eds.; World Scientific: Singapore, 2008; pp. 477–486. [Google Scholar]
- Kitony, J.K.; Sunohara, H.; Tasaki, M.; Mori, J.-I.; Shimazu, A.; Reyes, V.P.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an aus-derived nested association mapping (aus-NAM) population in rice. Plants 2021, 10, 1255. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, C.A.; Moreno, M.; Wang, Z.; Heffelfinger, C.; Arbelaez, L.J.; Aguirre, J.A.; Franco, N.; Romero, L.E.; Labadie, K.; Zhao, H.; et al. Genetic architecture of a rice nested association mapping population. G3 Genes Genomes Genet. 2017, 7, 1913–1926. [Google Scholar] [CrossRef]
- Bandillo, N.; Raghavan, C.; Muyco, P.; Sevilla, M.A.L.; Lobina, I.T.; Dilla-Ermita, C.; Tung, C.-W.; McCouch, S.; Thomson, M.; Mauleon, R.; et al. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: Progress and potential for genetics research and breeding. Rice 2013, 6, 11. [Google Scholar] [CrossRef]
- Ogawa, D.; Yamamoto, E.; Ohtani, T.; Kanno, N.; Tsunematsu, H.; Nonoue, Y.; Yano, M.; Yamamoto, T.; Yonemaru, J. Haplotype-based allele mining in the Japan-MAGIC rice population. Sci. Rep. 2018, 8, 4379. [Google Scholar] [CrossRef]
- Norton, G.J.; Travis, A.J.; Douglas, A.; Fairley, S.; Alves, E.D.P.; Ruang-areerate, P.; Naredo, M.E.B.; McNally, K.L.; Hossain, M.; Islam, M.R.; et al. Genome wide association mapping of grain and straw biomass traits in the rice Bengal and Assam aus panel (BAAP) grown under alternate wetting and drying and permanently flooded irrigation. Front. Plant Sci. 2018, 9, 1223. [Google Scholar] [CrossRef]
- Eizenga, G.C.; Ali, M.L.; Bryant, R.J.; Yeater, K.M.; McClung, A.M.; McCouch, S.R. Registration of the rice diversity panel 1 for genomewide association studies. J. Plant Regist. 2014, 8, 109–116. [Google Scholar] [CrossRef]
- Hoang, G.T.; Van Dinh, L.; Nguyen, T.T.; Ta, N.K.; Gathignol, F.; Mai, C.D.; Jouannic, S.; Tran, K.D.; Khuat, T.H.; Do, V.N.; et al. Genome-wide association study of a panel of Vietnamese rice landraces reveals new QTLs for tolerance to water deficit during the vegetative phase. Rice 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Gorjanc, G.; Jenko, J.; Hearne, S.J.; Hickey, J.M. Initiating maize pre-breeding programs using genomic selection to harness polygenic variation from landrace populations. BMC Genom. 2016, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sánchez-Villeda, H.; Sorrells, M.; et al. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genom. 2012, 11, 3–11. [Google Scholar] [CrossRef]
- Bentley, D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006, 16, 545–552. [Google Scholar] [CrossRef]
- Quail, M.; Smith, M.E.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A Tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid snp discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Van Tassell, C.P.; Smith, T.P.L.; Matukumalli, L.K.; Taylor, J.F.; Schnabel, R.D.; Lawley, C.T.; Haudenschild, C.D.; Moore, S.S.; Warren, W.C.; Sonstegard, T.S. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 2008, 5, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive Method for De Novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Truong, H.T.; Ramos, A.M.; Yalcin, F.; de Ruiter, M.; van der Poel, H.J.A.; Huvenaars, K.H.J.; Hogers, R.C.J.; van Enckevort, L.J.G.; Janssen, A.; van Orsouw, N.J.; et al. Sequence-based genotyping for marker discovery and co-dominant scoring in germplasm and populations. PLoS ONE 2012, 7, e37565. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, S.; Hirase, S.; Kikuchi, K.; Nanjo, K.; Nakamura, Y.; Kohno, H.; Sano, M. Random PCR-based genotyping by sequencing technology GRAS-Di (genotyping by random amplicon sequencing, direct) reveals genetic structure of mangrove fishes. Mol. Ecol. Resour. 2019, 19, 1153–1163. [Google Scholar] [CrossRef]
- Enoki, H. The construction of psedomolecules of a commercial strawberry by DeNovoMAGIC and new genotyping technology, GRAS-Di. In Proceedings of the Plant and Animal Genome Conference XXVII, San Diego, CA, USA, 12–16 January 2019. [Google Scholar]
- Enoki, H.; Takeuchi, Y. New genotyping technology, GRAS-Di, using next generation sequencer. In Proceedings of the Plant and Animal Genome Conference XXVI, San Diego, CA, USA, 13–17 January 2018. [Google Scholar]
- Suyama, Y.; Matsuki, Y. MIG-Seq: An effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing Platform. Sci. Rep. 2015, 5, 16963. [Google Scholar] [CrossRef]
- Onda, Y.; Takahagi, K.; Shimizu, M.; Inoue, K.; Mochida, K. Multiplex PCR targeted amplicon sequencing (MTA-Seq): Simple, flexible, and versatile SNP genotyping by highly multiplexed PCR amplicon sequencing. Front. Plant Sci. 2018, 9, 201. [Google Scholar] [CrossRef]
- Campbell, N.R.; Harmon, S.A.; Narum, S.R. Genotyping-in-thousands by sequencing (GT-seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 2015, 15, 855–867. [Google Scholar] [CrossRef]
- Telfer, E.; Graham, N.; Macdonald, L.; Li, Y.; Klápště, J.; Resende, M., Jr.; Neves, L.G.; Dungey, H.; Wilcox, P. A high-density exome capture genotype-by-sequencing panel for forestry breeding in Pinus radiata. PLoS ONE 2019, 14, e0222640. [Google Scholar] [CrossRef]
- Toonen, R.J.; Puritz, J.B.; Forsman, Z.H.; Whitney, J.L.; Fernandez-Silva, I.; Andrews, K.R.; Bird, C.E. EzRAD: A simplified method for genomic genotyping in non-model organisms. PeerJ 2013, 1, e203. [Google Scholar] [CrossRef]
- Nishimura, K.; Motoki, K.; Yamazaki, A.; Takisawa, R.; Yasui, Y.; Kawai, T.; Ushijima, K.; Nakano, R.; Nakazaki, T. MIG-Seq is an effective method for high-throughput genotyping in wheat (Triticum spp.). DNA Res. 2022, 29, dsac011. [Google Scholar] [CrossRef]
- Umeda, M.; Sakaigaichi, T.; Tanaka, M.; Tarumoto, Y.; Adachi, K.; Hattori, T.; Hayano, M.; Takahashi, H.; Tamura, Y.; Kimura, T.; et al. Detection of a major QTL related to smut disease resistance inherited from a Japanese wild sugarcane using GRAS-Di technology. Breed. Sci. 2021, 71, 365–374. [Google Scholar] [CrossRef]
- Kumawat, G.; Xu, D. A major and stable quantitative trait locus qSS2 for seed size and shape traits in a soybean RIL population. Front. Genet. 2021, 12, 646102. [Google Scholar] [CrossRef]
- Miki, Y.; Yoshida, K.; Enoki, H.; Komura, S.; Suzuki, K.; Inamori, M.; Nishijima, R.; Takumi, S. GRAS-Di system facilitates high-density genetic map construction and qtl identification in recombinant inbred lines of the wheat progenitor Aegilops tauschii. Sci. Rep. 2020, 10, 21455. [Google Scholar] [CrossRef]
- Suren, H.; Hodgins, K.A.; Yeaman, S.; Nurkowski, K.A.; Smets, P.; Rieseberg, L.H.; Aitken, S.N.; Holliday, J.A. Exome capture from the spruce and pine giga-genomes. Mol. Ecol. Resour. 2016, 16, 1136–1146. [Google Scholar] [CrossRef]
- Neves, L.G.; Davis, J.M.; Barbazuk, W.B.; Kirst, M. Whole-exome targeted sequencing of the uncharacterized pine genome. Plant J. 2013, 75, 146–156. [Google Scholar] [CrossRef]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef]
- Arbelaez, J.D.; Moreno, L.T.; Singh, N.; Tung, C.-W.; Maron, L.G.; Ospina, Y.; Martinez, C.P.; Grenier, C.; Lorieux, M.; McCouch, S. Development and GBS-genotyping of introgression lines (ILs) using two wild species of rice, O. meridionalis and O. rufipogon, in a common recurrent parent, O. sativa cv. Curinga. Mol. Breed. 2015, 35, 81. [Google Scholar] [CrossRef]
- Spindel, J.; Wright, M.; Chen, C.; Cobb, J.; Gage, J.; Harrington, S.; Lorieux, M.; Ahmadi, N.; McCouch, S. Bridging the genotyping gap: Using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 2013, 126, 2699–2716. [Google Scholar] [CrossRef]
- Furuta, T.; Ashikari, M.; Jena, K.K.; Doi, K.; Reuscher, S. Adapting genotyping-by-sequencing for rice F2 populations. G3 Genes Genomes Genet. 2017, 7, 881–893. [Google Scholar] [CrossRef]
- Reyes, V.P.; Angeles-Shim, R.B.; Mendioro, M.S.; Manuel, M.C.C.; Lapis, R.S.; Shim, J.; Sunohara, H.; Nishiuchi, S.; Kikuta, M.; Makihara, D.; et al. Marker-assisted introgression and stacking of major QTLs controlling grain number (Gn1a) and number of primary branching (WFP) to NERICA Cultivars. Plants 2021, 10, 844. [Google Scholar] [CrossRef]
- Soe, T.K.; Kunieda, M.; Sunohara, H.; Inukai, Y.; Reyes, V.P.; Nishiuchi, S.; Doi, K. A novel combination of genes causing temperature-sensitive hybrid weakness in rice. Front. Plant Sci. 2022, 13, 908000. [Google Scholar] [CrossRef]
- Yamada, S.; Kurokawa, Y.; Nagai, K.; Angeles-Shim, R.B.; Yasui, H.; Furuya, N.; Yoshimura, A.; Doi, K.; Ashikari, M.; Sunohara, H. Evaluation of backcrossed pyramiding lines of the yield-related gene and the bacterial leaf blight resistant genes. J. Intl. Coop. Agric. Dev. 2020, 18, 18–28. [Google Scholar]
- Liang, Y.; Dong, X.; Ni, X.; Wang, Q.; Sahu, S.K.; Hou, J.; Liang, M.; Chen, L.; Zhang, G. Genotyping by sequencing of 270 indica rice varieties revealed genetic markers probably related to heavy metal accumulation. Plant Breed. 2018, 137, 691–697. [Google Scholar] [CrossRef]
- Goto, I.; Neang, S.; Kuroki, R.; Reyes, V.P.; Doi, K.; Skoulding, N.S.; Taniguchi, M.; Yamauchi, A.; Mitsuya, S. QTL analysis for sodium removal ability in rice leaf sheaths under salinity using an IR-44595/318 F2 population. Front. Plant Sci. 2022, 13, 1002605. [Google Scholar] [CrossRef] [PubMed]
- Waheed, R.; Ignacio, J.C.; Arbelaez, J.D.; Juanillas, V.M.; Asif, M.; Henry, A.; Kretzschmar, T.; Arif, M. Drought response QTLs in a super basmati × azucena population by high-density GBS-based SNP linkage mapping. Plant Breed. 2021, 140, 758–774. [Google Scholar] [CrossRef]
- Suman, K.; Neeraja, C.N.; Madhubabu, P.; Rathod, S.; Bej, S.; Jadhav, K.P.; Kumar, J.A.; Chaitanya, U.; Pawar, S.C.; Rani, S.H.; et al. Identification of promising RILs for high grain zinc through genotype × environment analysis and stable grain zinc QTL using SSRs and SNPs in rice (Oryza sativa L.). Front. Plant Sci. 2021, 12, 587482. [Google Scholar] [CrossRef]
- De Leon, T.B.; Linscombe, S.; Subudhi, P.K. Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) using a high-density GBS-based SNP linkage map. Rice 2016, 9, 52. [Google Scholar] [CrossRef]
- Chen, L.; Gao, W.; Chen, S.; Wang, L.; Zou, J.; Liu, Y.; Wang, H.; Chen, Z.; Guo, T. High-resolution QTL mapping for grain appearance traits and co-localization of chalkiness-associated differentially expressed candidate genes in rice. Rice 2016, 9, 48. [Google Scholar] [CrossRef]
- Bernardo, R. Genomewide selection for rapid introgression of exotic germplasm in maize. Crop Sci. 2009, 49, 419–425. [Google Scholar] [CrossRef]
- Combs, E.; Bernardo, R. Genomewide selection to introgress semidwarf maize germplasm into U.S. corn belt inbreds. Crop Sci. 2013, 53, 1427–1436. [Google Scholar] [CrossRef]
- Grenier, C.; Cao, T.-V.; Ospina, Y.; Quintero, C.; Châtel, M.H.; Tohme, J.; Courtois, B.; Ahmadi, N. Accuracy of genomic selection in a rice synthetic population developed for recurrent selection breeding. PLoS ONE 2015, 10, e0136594. [Google Scholar] [CrossRef]
- Marulanda, J.J.; Mi, X.; Melchinger, A.E.; Xu, J.-L.; Würschum, T.; Longin, C.F.H. Optimum breeding strategies using genomic selection for hybrid breeding in wheat, maize, rye, barley, rice and triticale. Theor. Appl. Genet. 2016, 129, 1901–1913. [Google Scholar] [CrossRef]
- Onogi, A.; Ideta, O.; Inoshita, Y.; Ebana, K.; Yoshioka, T.; Yamasaki, M.; Iwata, H. Exploring the areas of applicability of whole-genome prediction methods for asian rice (Oryza sativa L.). Theor. Appl. Genet. 2015, 128, 41–53. [Google Scholar] [CrossRef]
- Onogi, A.; Watanabe, M.; Mochizuki, T.; Hayashi, T.; Nakagawa, H.; Hasegawa, T.; Iwata, H. Toward integration of genomic selection with crop modelling: The development of an integrated approach to predicting rice heading dates. Theor. Appl. Genet. 2016, 129, 805–817. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef]
- Spindel, J.E.; Begum, H.; Akdemir, D.; Collard, B.; Redoña, E.; Jannink, J.-L.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef]
- Wimmer, V.; Lehermeier, C.; Albrecht, T.; Auinger, H.-J.; Wang, Y.; Schön, C.-C. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics 2013, 195, 573–587. [Google Scholar] [CrossRef]
- Monteverde, E.; Rosas, J.E.; Blanco, P.; Pérez de Vida, F.; Bonnecarrère, V.; Quero, G.; Gutierrez, L.; McCouch, S. Multienvironment models increase prediction accuracy of complex traits in advanced breeding lines of rice. Crop Sci. 2018, 58, 1519–1530. [Google Scholar] [CrossRef]
- Bhandari, A.; Bartholomé, J.; Cao-Hamadoun, T.-V.; Kumari, N.; Frouin, J.; Kumar, A.; Ahmadi, N. Selection of trait-specific markers and multi-environment models improve genomic predictive ability in rice. PLoS ONE 2019, 14, e0208871. [Google Scholar] [CrossRef]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.-L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Phocas, F. Genotyping, the usefulness of imputation to increase SNP density, and imputation methods and tools. In Genomic Prediction of Complex Traits; Ahmadi, N., Bartholomé, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2022; Volume 2467, pp. 113–138. [Google Scholar]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Nazzicari, N.; Biscarini, F.; Cozzi, P.; Brummer, E.C.; Annicchiarico, P. Marker imputation efficiency for genotyping-by-sequencing data in rice (Oryza sativa) and Alfalfa (Medicago sativa). Mol. Breed. 2016, 36, 69. [Google Scholar] [CrossRef]
- Lorieux, M.; Gkanogiannis, A.; Fragoso, C.; Rami, J.-F. NOISYmputer: Genotype imputation in bi-parental populations for noisy low-coverage next-generation sequencing data. bioRxiv 2019. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-Generation DNA Sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef]
- Wickland, D.P.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-EaSy. BMC Bioinform. 2017, 18, 586. [Google Scholar] [CrossRef]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Légaré, G.; Boyle, B.; Normandeau, É.; Laroche, J.; Larose, S.; Jean, M.; et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Laroche, J.; Bastien, M.; Abed, A.; Belzile, F. Fast-GBS: A new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinform. 2017, 18, 5. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef]
- Reyes, V.P. Application of Next-Generation Sequencing Technology for Genetic Analysis and Pre-Breeding of Rice. Ph.D. Dissertation, Nagoya University, Nagoya, Japan, 2021. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Type | Advantages | Disadvantages | Examples | References |
|---|---|---|---|---|---|
| Restriction enzyme-based | Single enzyme |
| RRL, RAD-seq, Elshire’s GBS, | [20,21,22] | |
| Double enzyme | Provides a greater degree of complexity reduction |
| ddRAD, Poland’s GBS, SGB, eZRAD | [23,24,25] | |
| PCR-based | Random |
|
| Genotyping by random amplicon sequencing, direct (GRAS-Di); MIG-Seq | [26,27,28] |
| Targeted | GT-seq; MTA-Seq; | [29,30,31] | |||
| Target capture |
|
| Exon capture; Capture of known polymorphic sites | [32] |
| Trait | QTL | Type of Population | Reference |
|---|---|---|---|
| Hybrid weakness | hwj1 and hwj2 | F2 | [45] |
| Shoot Na+ concentration | qSNC1-1, qSNC1-2, and qSNC11 | F2 | [48] |
| Leaf sheath Na+ concentration | qSHNC1 and qSHNC11 | ||
| Leaf blade K+ -Na+ ratio | qBKNR11 | ||
| Water uptake | qWU7 and qWU11 | RIL | [49] |
| Zinc content in polished rice | qZPR1.1 | BRIL | [50] |
| Salinity Tolerance | qSIS5.1b and qSIS6.30 | RIL | [51] |
| Grain quality | qGS5.2, qGS7.1, and qPGWC8 | RIL | [52] |
| GBS-Pipeline | SNP Calling Strategy | Ease of Use 1 | Reference |
|---|---|---|---|
| Trait Analysis by aSSociation, Evolution and Linkage (TASSEL) | Binomial likelihood ratio | Needs extra steps to improve SNP call accuracy. Not built into the pipeline | [68] |
| IBIS genotyping by sequencing tools (IGST) | Bayesian | - | [76] |
| Fast-GBS | Haplotype-based | Needs extra steps to improve SNP call accuracy. Not built into the pipeline | [77] |
| Stacks | Multinomial-based likelihood | - | [78] |
| GB-eaSy | Bayesian | Additional steps for SNP call accuracy is not needed or built into the pipeline itself. | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, V.P.; Kitony, J.K.; Nishiuchi, S.; Makihara, D.; Doi, K. Utilization of Genotyping-by-Sequencing (GBS) for Rice Pre-Breeding and Improvement: A Review. Life 2022, 12, 1752. https://doi.org/10.3390/life12111752
Reyes VP, Kitony JK, Nishiuchi S, Makihara D, Doi K. Utilization of Genotyping-by-Sequencing (GBS) for Rice Pre-Breeding and Improvement: A Review. Life. 2022; 12(11):1752. https://doi.org/10.3390/life12111752
Chicago/Turabian StyleReyes, Vincent Pamugas, Justine Kipruto Kitony, Shunsaku Nishiuchi, Daigo Makihara, and Kazuyuki Doi. 2022. "Utilization of Genotyping-by-Sequencing (GBS) for Rice Pre-Breeding and Improvement: A Review" Life 12, no. 11: 1752. https://doi.org/10.3390/life12111752
APA StyleReyes, V. P., Kitony, J. K., Nishiuchi, S., Makihara, D., & Doi, K. (2022). Utilization of Genotyping-by-Sequencing (GBS) for Rice Pre-Breeding and Improvement: A Review. Life, 12(11), 1752. https://doi.org/10.3390/life12111752