
Inferring Therapeutic Targets in Candida albicans and Possible Inhibition through Natural Products: A Binding and Physiological Based Pharmacokinetics Snapshot
 ,
,  ,
,
Abstract
1. Introduction
2. Material & Methods
2.1. Data Retrieval
2.2. Essentiality Analysis
2.3. Drug Target Mining
2.4. Virtual Screening
2.5. ADMET Profiling
3. Results
3.1. Therapeutic Candidate Mining
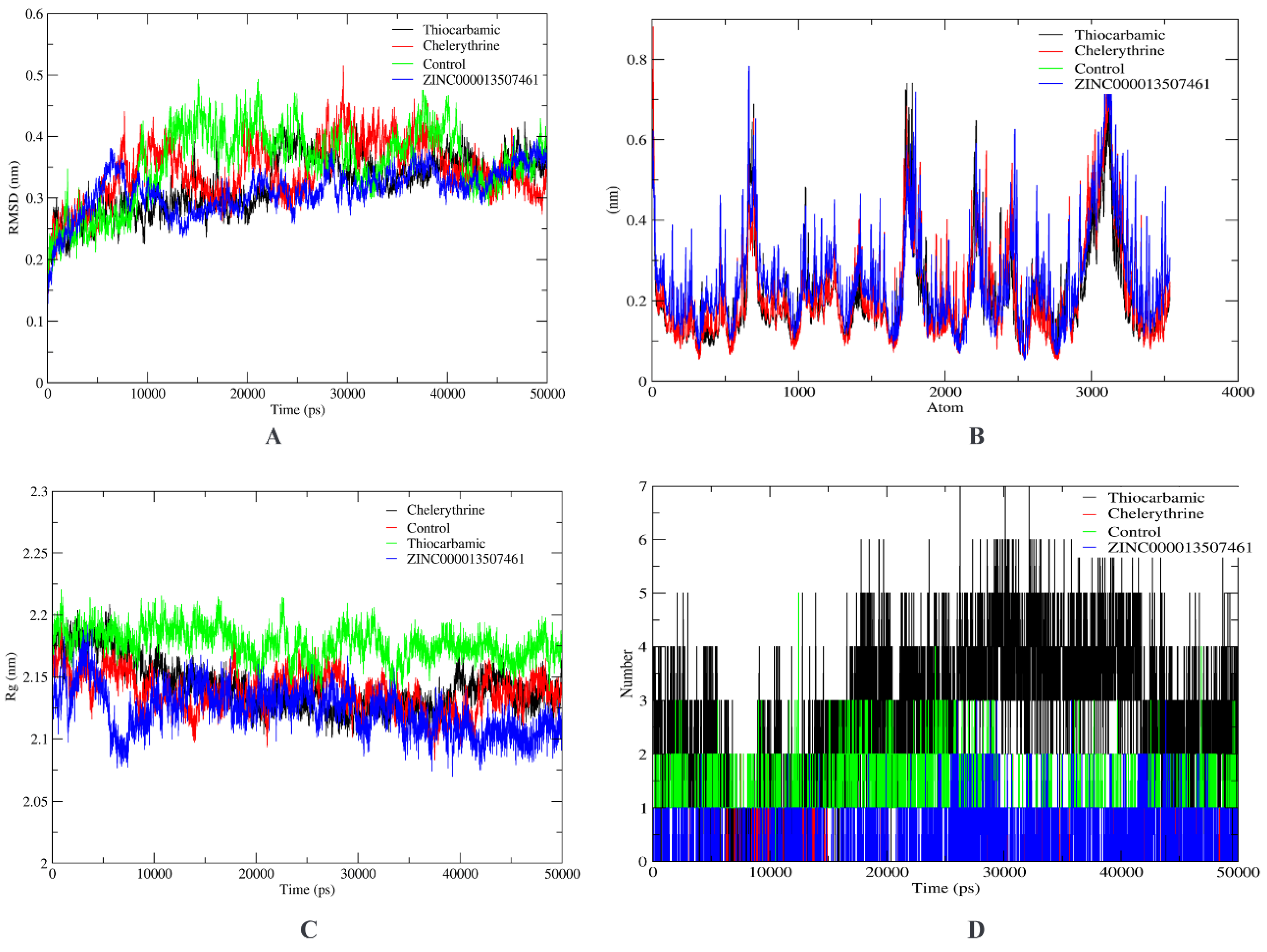
3.2. Virtual Screening
3.3. ADMET Profiling
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Achkar, J.M.; Fries, B.C. Candida infections of the genitourinary tract. Clin. Microbiol. Rev. 2010, 23, 253–273. [Google Scholar] [CrossRef]
- Pellon, A.; Begum, N.; Sadeghi Nasab, S.D.; Harzandi, A.; Shoaie, S.; Moyes, D.L. Role of Cellular Metabolism during Candida-Host Interactions. Pathogens 2022, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.R.; Gaffen, S.L.; Hube, B. Candidalysin: Discovery and function in Candida albicans infections. Curr. Opin. Microbiol. 2019, 52, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Roy, A.; Singh, S.; Pradhan, P.; Garg, P.; Singh, N. Factors determining the mortality in cirrhosis patients with invasive candidiasis: A systematic review and meta-analysis. Med. Mycol. 2021, 60, myab069. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Patterson, T.F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J. Infect. Dis. 2017, 216 (Suppl. 3), S445–S451. [Google Scholar] [CrossRef]
- Healey, K.R.; Perlin, D.S. Fungal resistance to echinocandins and the MDR phenomenon in Candida glabrata. J. Fungi 2018, 4, 105. [Google Scholar] [CrossRef]
- Zhang, Y.; Muend, S.; Rao, R. Dysregulation of ion homeostasis by antifungal agents. Front. Microbiol. 2012, 3, 133. [Google Scholar] [CrossRef]
- Silva, S.; Rodrigues, C.F.; Araújo, D.; Rodrigues, M.E.; Henriques, M. Candida species biofilms’ antifungal resistance. J. Fungi 2017, 3, 8. [Google Scholar] [CrossRef]
- Spampinato, C.; Leonardi, D. Candida Infections, Causes, Targets, and Resistance Mechanisms: Traditional and Alternative Antifungal Agents. BioMed Res. Int. 2013, 2013, 204237. [Google Scholar] [CrossRef]
- Spettel, K.; Barousch, W.; Makristathis, A.; Zeller, I.; Nehr, M.; Selitsch, B.; Lackner, M.; Rath, P.; Steinmann, J.; Willinger, B. Analysis of antifungal resistance genes in Candida albicans and Candida glabrata using next generation sequencing. PLoS ONE 2019, 14, e0210397. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Spaltmann, F.; Blunck, M.; Ziegelbauer, K. Computer-aided target selection—Prioritizing targets for antifungal drug discovery. Drug Discov. Today 1999, 4, 17–26. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Luo, H.; Lin, Y.; Liu, T.; Lai, F.; Zhang, C.; Gao, F.; Zhang, R. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 2021, 49, D677–D686. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.N.; Hua, Z.G.; Huang, J.; Rao, N.; Guo, F.B. CEG: A database of essential gene clusters. BMC Genom. 2013, 14, 769. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.-X.; Liu, W.; Wang, C.; Zhang, F.; Ye, Y.; Wu, C.; Zheng, W.; Rao, N.; Guo, F. CEG 2.0: An updated database of clusters of essential genes including eukaryotic organisms. Database 2020, 2020, baaa112. [Google Scholar] [CrossRef]
- Glass, J.I.; Assad-Garcia, N.; Alperovich, N.; Yooseph, S.; Lewis, M.R.; Maruf, M.; Hutchison, C.A.; Smith, H.O.; Venter, J.C. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 2006, 103, 425–430. [Google Scholar] [CrossRef]
- Basharat, Z.; Jahanzaib, M.; Rahman, N. Therapeutic target identification via differential genome analysis of antibiotic resistant Shigella sonnei and inhibitor evaluation against a selected drug target. Infect. Genet. Evol. 2021, 94, 105004. [Google Scholar] [CrossRef]
- Basharat, Z.; Jahanzaib, M.; Yasmin, A.; Khan, I.A. Pan-genomics, drug candidate mining and ADMET profiling of natural product inhibitors screened against Yersinia pseudotuberculosis. Genomics 2021, 113, 238–244. [Google Scholar] [CrossRef]
- Nasim, F.; Dey, A.; Qureshi, I.A. Comparative genome analysis of Corynebacterium species: The underestimated pathogens with high virulence potential. Infect. Genet. Evol. 2021, 93, 104928. [Google Scholar] [CrossRef]
- Basharat, Z.; Akhtar, U.; Khan, K.; Alotaibi, G.; Jalal, K.; Abbas, M.N.; Hayat, A.; Ahmad, D.; Hassan, S.S. Differential analysis of Orientia tsutsugamushi genomes for therapeutic target identification and possible intervention through natural product inhibitor screening. Comput. Biol. Med. 2022, 141, 105165. [Google Scholar] [CrossRef] [PubMed]
- Chakkyarath, V.; Shanmugam, A.; Natarajan, J. Prioritization of potential drug targets and antigenic vaccine candidates against Klebsiella aerogenes using the computational subtractive proteome-driven approach. J. Proteins Proteom. 2021, 12, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Basharat, Z.; Jalal, K.; Mashraqi, M.M.; Alzamami, A.; Alshamrani, S.; Uddin, R. Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products. Antibiotics 2022, 11, 680. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhu, X.; Hong, Z.; Wei, L.; Ren, Y.; Wan, F.; Zhu, S.; Peng, H.; Guo, L.; Rao, L.; et al. Structure-Based Rational Design of Novel Inhibitors Against Fructose-1,6-Bisphosphate Aldolase from Candida albicans. J. Chem. Inf. Model. 2017, 57, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Jalal, K.; Abu-Izneid, T.; Khan, K.; Abbas, M.; Hayat, A.; Bawazeer, S.; Uddin, R. Identification of vaccine and drug targets in Shigella dysenteriae sd197 using reverse vaccinology approach. Sci. Rep. 2022, 12, 251. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Hussain, A.; Altamimi, M.A.; Afzal, O.; Altamimi, A.S.A.; Ali, A.; Martinez, F.; Siddique, M.U.M.; Acree, W.E., Jr.; Jouyban, A. Preferential Solvation Study of the Synthesized Aldose Reductase Inhibitor (SE415) in the {PEG 400 (1) + Water (2)} Cosolvent Mixture and GastroPlus-Based Prediction. ACS Omega 2022, 7, 1197–1210. [Google Scholar] [CrossRef]
- Talapphetsakun, T.; Viyoch, J.; Waranuch, N.; Sermsappasuk, P. The Development of a Physiologically Based Pharmacokinetic (PBPK) Model of Andrographolide in Mice and Scaling It up to Rats, Dogs and Humans. Curr. Drug Metab. 2022, 23, 15. [Google Scholar] [CrossRef]
- Rodaki, A.; Young, T.; Brown, A.J. Effects of depleting the essential central metabolic enzyme fructose-1,6-bisphosphate aldolase on the growth and viability of Candida albicans: Implications for antifungal drug target discovery. Eukaryot. Cell 2006, 5, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Nitia, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- d’Enfert, C.; Kaune, A.-K.; Alaban, L.-R.; Chakraborty, S.; Cole, N.; Delavy, M.; Kosmala, D.; Marsaux, B.; Frois-Martins, R.; Morelli, M.; et al. The impact of the Fungus-Host-Microbiota interplay upon Candida albicans infections: Current knowledge and new perspectives. FEMS Microbiol. Rev. 2021, 45, fuaa060. [Google Scholar] [CrossRef] [PubMed]
- Köhler, J.R.; Casadevall, A.; Perfect, J. The spectrum of fungi that infects humans. Cold Spring Harb. Perspect. Med. 2015, 5, a019273. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liao, K.; Wang, D. Effects of magnolol and honokiol on adhesion, yeast-hyphal transition, and formation of biofilm by Candida albicans. PLoS ONE 2015, 10, e0117695. [Google Scholar] [CrossRef]
- Li, Y.; Sun, L.; Lu, C.; Gong, Y.; Li, M.; Sun, S. Promising antifungal targets against Candida albicans based on ion homeostasis. Front. Cell. Infect. Microbiol. 2018, 8, 286. [Google Scholar] [CrossRef]
- Vila, T.; Romo, J.A.; Pierce, C.G.; McHardy, S.F.; Saville, S.P.; Lopez-Ribot, J.L. Targeting Candida albicans filamentation for antifungal drug development. Virulence 2017, 8, 150–158. [Google Scholar] [CrossRef]
- Guo, X.L.; Leng, P.; Yang, Y.; Luo, H.X. Plagiochin E, a botanic-derived phenolic compound, reverses fungal resistance to fluconazole relating to the efflux pump. J. Appl. Microbiol. 2008, 104, 831–838. [Google Scholar] [CrossRef]
- Khan, K.; Jalal, K.; Khan, A.; Al-Harrasi, A.; Uddin, R. Comparative Metabolic Pathways Analysis and Subtractive Genomics Profiling to Prioritize Potential Drug Targets Against Streptococcus pneumoniae. Front. Microbiol. 2021, 12, 796363. [Google Scholar] [CrossRef]
- Bappy, M.N.I.; Robin, T.B.; Prome, A.A.; Laskar, F.S.; Roy, A.; Akter, H.; Zinnah, K.M.A. Subtractive proteomics analysis to uncover the potent drug targets for distinctive drug design of Candida auris. bioRxiv 2022. [Google Scholar] [CrossRef]
- de Amorim, A.L.; de Lima, A.V.M.; Rosário, A.; Souza, E.T.D.S.; Ferriera, J.V.; Hage-Melim, L.I.dS. Molecular modeling of inhibitors against fructose bisphosphate aldolase from Candida albicans. Silico Pharmacol. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Elamin Elhasan, L.M.; Hassan, M.B.; Elhassan, R.M.; Abdelrhman, F.A.; Salih, E.A.; Ibrahim, A.; Mohamed, A.A.; Osman, H.S.; Khalil, M.S.M.; Alsafi, A.A.; et al. Epitope-based peptide vaccine design against fructose bisphosphate aldolase of Candida glabrata: An immunoinformatics approach. J. Immunol. Res. 2021, 2021, 8280925. [Google Scholar] [CrossRef] [PubMed]
- Rodicio, R.; Schmitz, H.-P.; Heinisch, J.J. Genetic and physiological characterization of fructose-1, 6-bisphosphate aldolase and glyceraldehyde-3-phosphate dehydrogenase in the crabtree-negative yeast Kluyveromyces lactis. Int. J. Mol. Sci. 2022, 23, 772. [Google Scholar] [CrossRef] [PubMed]
- Pirovich, D.B.; Da’dara, A.A.; Skelly, P.J. Multifunctional fructose 1, 6-bisphosphate aldolase as a therapeutic target. Front. Mol. Biosci. 2021, 8, 719678. [Google Scholar] [CrossRef] [PubMed]
- Zavrel, M.; White, T.C. Medically important fungi respond to azole drugs: An update. Future Microbiol. 2015, 10, 1355–1373. [Google Scholar] [CrossRef] [PubMed]
- Jalal, K.; Khan, K.; Hassam, M.; Abbas, M.N.; Uddin, R.; Khusro, A.; Sahibzada, M.U.K.; Gajdacs, M. Identification of a Novel Therapeutic Target against XDR Salmonella Typhi H58 Using Genomics Driven Approach Followed up by Natural Products Virtual Screening. Microorganisms 2021, 9, 2512. [Google Scholar] [CrossRef]
- Wangyal, R.; Tidwell, T.; Dhondrup, W.; Yungdrung, T.; Dhondrup, G.; He, Q.; Zhang, Y. Dataset of materia medica in Sowa Rigpa: Tibetan medicine botanicals and Gawé Dorjé’s classification system. Data Brief 2020, 33, 106498. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K. Indian traditional ayurvedic system of medicine and nutritional supplementation. Evid.Based Complement. Altern. Med. 2013, 2013, 376327. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.J.; Xu, T.; Du, H.; Huan Gang, C.L.; Huan Gang, C.L.; Fan, G.; Zhang, Y. Natural Medicines Used in the Traditional Tibetan Medical System for the Treatment of Liver Diseases. Front. Pharmacol. 2018, 9, 29. [Google Scholar] [CrossRef]
- Devpura, G.; Tomar, B.S.; Nathiya, D.; Sharma, A.; Bhandari, D.; Haldar, S.; Balkrishna, A.; Varshney, A. Randomized placebo-controlled pilot clinical trial on the efficacy of ayurvedic treatment regime on COVID-19 positive patients. Phytomedicine 2021, 84, 153494. [Google Scholar] [CrossRef]
- Liu, W.; Wu, Y.H.; Hu, S.Y.; Zhong, C.; Gao, M.; Liu, D.; Wang, H.; Chen, M.; Song, Y.; Yang, B.; et al. A multicenter, randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of Tong Luo Hua Shi capsule, a modernized Tibetan medicine, in patients with rheumatoid arthritis. Trials 2016, 17, 359. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.X.; Dong, X.; Xie, Z.M.; Li, X.; Wang, X.; Huang, J.; Wei, S.; Liu, Y.; Liu, J. Efficacy and safety of Tibetan medicine Qingpeng ointment for acute gouty arthritis: Protocol for a multi-center, randomized, double-blind, placebo-controlled trial. Trials 2022, 23, 387. [Google Scholar] [CrossRef] [PubMed]
- Witt, C.M.; Michalsen, A.; Roll, S.; Morandi, A.; Gupta, S.; Rosenberg, M.; Kronpass, L.; Stapelfeldt, E.; Hisar, S.; Muller, M.; et al. Comparative effectiveness of a complex Ayurvedic treatment and conventional standard care in osteoarthritis of the knee-study protocol for a randomized controlled trial. Trials 2013, 14, 149. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Hoseini-Alfatemi, S.M.; Sharifi-Rad, M.; Sahrifi-Rad, M.; Iriti, M.; Sharifi-Rad, M.; Sharifi-Rad, R.; Raeisis, S. Phytochemical Compositions Biological Activities of Essential Oil from Xanthium strumarium L. Biomolecules 2015, 20, 7034–7047. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Soufi, L.; Ayatollahi, S.A.; Iriti, M.; Sharifi-Rad, M.; Varoni, E.M.; Shahri, F.; Esposito, S.; Kuhestani, K.; Sharifi-Rad, M. Anti-bacterial effect of essential oil from Xanthium strumarium against shiga toxin-producing Escherichia coli. Cell. Mol. Biol. 2016, 62, 69–74. [Google Scholar]
- Soulaimani, B.; Varoni, E.; Iriti, M.; Mezrioui, N.; Hassani, L.; Abbad, A. Synergistic Anticandidal Effects of Six Essential Oils in Combination with Fluconazole or Amphotericin B against Four Clinically Isolated Candida Strains. Antibiotics 2021, 10, 1049. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the h ACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Brogi, S.J.M. Computational approaches for drug discovery. Molecules 2019, 24, 3061. [Google Scholar] [CrossRef]
- Yasmin, A.; Basharat, Z.; Safdar, N. In-silico Approach to Target Cancer Cell DNA Repair Pathway. In Phytochemistry: An In-Silico and In-Vitro Update; Springer: Berlin/Heidelberg, Germany, 2019; pp. 373–392. [Google Scholar]
- Herbert, J.; Augereau, J.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent specific inhibitor of protein kinase, C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Fan, L.; Fan, Y.; Liu, L.; Tao, W.; Shan, X.; Dong, Y.; Li, L.; Zhang, S.; Wang, H. Chelerythrine attenuates the inflammation of lipopolysaccharide-induced acute lung inflammation through NF-κB signaling pathway mediated by Nrf2. Front. Pharmacol. 2018, 9, 1047. [Google Scholar] [CrossRef]
- De Buck, S.S.; Sinha, V.K.; Fenu, L.A.; Nijsen, M.J.; Mackie, C.E.; Gilissen, R.A.H.J. Prediction of human pharmacokinetics using physiologically based modeling: A retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispos. 2007, 35, 1766–1780. [Google Scholar] [CrossRef] [PubMed]
- Shiran, M.; Proctor, N.; Howgate, E.; Rowland-Yew, K.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of metabolic drug clearance in humans: In vitro–in vivo extrapolation vs. allometric scaling. Xenobiotica 2006, 36, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Parrott, N.; Lukacova, V.; Fraczkiewicz, G.; Bolger, M.B. Predicting pharmacokinetics of drugs using physiologically based modeling—Application to food effects. AAPS J. 2009, 11, 45–53. [Google Scholar] [CrossRef]
- Jones, H.M.; Parrott, N.; Ohlenbusch, G.; Lave, T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin. Pharmacokinet. 2006, 45, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Parrott, N.; Lave, T. Applications of physiologically based absorption models in drug discovery and development. Mol. Pharm. 2008, 5, 760–775. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Protein Accession Number | Protein Name | Protein Length | DrugBank Alignment Length | E-Value | Subcellular Localization |
|---|---|---|---|---|---|---|
| 1 | XP_019330652.1 | lumazine synthase | 206 | 157 | 1.33396 × 10−29 | Cytoplasmic |
| 2 | XP_019330750.1 | 3-deoxy−7-phosphoheptulonate synthase | 371 | 356 | 5.95566 × 10−127 | Cytoplasmic |
| 3 | XP_019330821.1 | trehalose 6-phosphate synthase/phosphatase complex subunit | 1007 | 383 | 1.09471 × 10−38 | Nuclear/Plasma membrane |
| 4 | XP_019331058.1 | anthranilate synthase | 522 | 410 | 3.47489 × 10−58 | Cytoplasmic |
| 5 | XP_019331115.1 | Bgl22p | 924 | 358 | 1.55548 × 10−12 | Cytoplasmic |
| 6 | XP_710092.2 | 4-amino-4-deoxychorismate synthase | 822 | 461 | 1.1003 × 10−36 | Nuclear |
| 7 | XP_710211.2 | bifunctional chorismate synthase/riboflavin reductase [NAD(P)H] | 413 | 376 | 1.39864 × 10−52 | Mitochondrial/Nuclear |
| 8 | XP_710312.1 | tryptophan synthase | 702 | 394 | 1.85163 × 10−144 | Cytoplasmic |
| 9 | XP_710700.2 | pantoate--beta-alanine ligase | 316 | 305 | 1.03048 × 10−68 | Nuclear |
| 10 | XP_710729.1 | 3-deoxy−7-phosphoheptulonate synthase | 370 | 353 | 3.03778 × 10−117 | Cytoplasmic/Nuclear |
| 11 | XP_711703.1 | hypothetical protein CAALFM_CR05750WA | 342 | 129 | 5.37008 × 10−0.8 | Cytoplasmic |
| 12 | XP_711706.1 | alpha, alpha-trehalose-phosphate synthase (UDP-forming) TPS1 | 478 | 472 | 3.18455 × 10−92 | Cytoplasmic |
| 13 | XP_712232.1 | isocitrate lyase 1 | 550 | 250 | 2.64563 × 10−44 | Peroxisomal |
| 14 | XP_713033.1 | sulfonate dioxygenase | 386 | 289 | 1.79648 × 10−24 | Nuclear/Cytoplasmic |
| 15 | XP_713320.2 | trifunctional histidinol dehydrogenase/phosphoribosyl-AMP cyclohydrolase/phosphoribosyl-ATP diphosphatase | 838 | 424 | 9.87896 × 10−114 | Cytoplasmic |
| 16 | XP_713806.1 | hypothetical protein CAALFM_C111290WA | 369 | 261 | 4.84039 × 10−35 | Cytoplasmic |
| 17 | XP_714207.2 | trifunctional dihydropteroate synthetase/dihydrohydroxymethylpterin pyrophosphokinase/dihydroneopterin aldolase | 829 | 788 | 3.71427 × 10−161 | Nuclear/Cytoplasmic |
| 18 | XP_714543.2 | hypothetical protein CAALFM_C209810CA | 434 | 400 | 1.30711 × 10−42 | Cytoplasmic |
| 19 | XP_714705.1 | hypothetical protein CAALFM_C305640WA | 425 | 308 | 2.28143 × 10−35 | Cytoplasmic |
| 20 | XP_714872.2 | Dqd1p | 146 | 124 | 4.79446 × 10−33 | Cytoplasmic |
| 21 | XP_715352.2 | uroporphyrinogen-III C-methyltransferase | 561 | 418 | 2.52115 × 10−42 | Nuclear/Cytoplasmic |
| 22 | XP_715357.1 | Ebp7p | 392 | 385 | 3.40361 × 10−70 | Cytoplasmic |
| 23 | XP_715408.1 | anthranilate phosphoribosyltransferase | 369 | 313 | 2.82806 × 10−41 | Cytoplasmic |
| 24 | XP_715440.2 | Oye32p | 432 | 379 | 4.31391 × 10−35 | Cytoplasmic |
| 25 | XP_715739.1 | dihydroorotase | 358 | 356 | 4.16856 × 10−54 | Cytoplasmic |
| 26 | XP_716238.1 | hypothetical protein CAALFM_CR08310CA | 385 | 286 | 2.24051 × 10−36 | Nuclear |
| 27 | XP_716751.1 | Hypothetical protein CAALFM_C601400WA | 676 | 419 | 5.7776 × 10−14 | Plasma membrane |
| 28 | XP_717003.2 | Nik1p | 1081 | 227 | 7.42174 × 10−20 | Nuclear/Cytoplasmic |
| 29 | XP_718052.2 | Ymx6p | 622 | 304 | 4.11558 × 10−0.6 | Plasma membrane |
| 30 | XP_718069.2 | phenylacrylic acid decarboxylase | 229 | 185 | 7.40789 × 10−68 | Plasma membrane |
| 31 | XP_718219.1 | 5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase | 775 | 767 | 0 | Cytoplasmic |
| 32 | XP_718255.2 | dethiobiotin synthase | 212 | 205 | 7.64158 × 10−16 | Chloroplast/cytoplasmic |
| 33 | XP_718258.2 | biotin synthase | 374 | 323 | 1.05435 × 10−100 | Mitochondrial |
| 34 | XP_719019.1 | 3-methyl-2-oxobutanoate hydroxymethyltransferase | 309 | 262 | 1.03531 × 10−55 | Mitochondrial |
| 35 | XP_719048.1 | 2-isopropylmalate synthase | 579 | 603 | 5.36732 × 10−170 | Cytoplasmic |
| 36 | XP_719116.2 | L-methionine (R)-S-oxide reductase | 175 | 134 | 2.56642 × 10−30 | Cytoplasmic |
| 37 | XP_721010.2 | trifunctional hydroxymethylpyrimidine kinase/phosphomethylpyrimidine kinase/thiaminase | 548 | 273 | 2.42294 × 10−26 | Cytoplasmic |
| 38 | XP_721446.1 | pyridoxine biosynthesis protein | 292 | 285 | 9.51495 × 10−106 | Cytoplasmic |
| 39 | XP_721536.1 | trehalose-phosphatase | 888 | 385 | 1.08126 × 10−54 | Cytoplasmic |
| 40 | XP_721716.2 | hypothetical protein CAALFM_C302070CA | 388 | 287 | 5.53839 × 10−34 | Cytoplasmic |
| 41 | XP_721934.1 | ATP phosphoribosyltransferase | 298 | 298 | 3.46987 × 10−33 | Cytoplasmic |
| 42 | XP_721932.2 | riboflavin synthase | 237 | 219 | 5.41917 × 10−35 | Cytoplasmic |
| 43 | XP_722690.1 | fructose-bisphosphate aldolase | 359 | 343 | 2.15136 × 10−129 | Cytoplasmic |
| 44 | XP_722769.2 | Aro1p | 1551 | 430 | 2.38324 × 10−70 | Cytoplasmic |
| 45 | XP_723161.2 | trifunctional fatty acid synthase sub-unit | 1884 | 763 | 2.04704 × 10−107 | Cytoplasmic |
| 46 | XP_723517.1 | Mts1p | 513 | 290 | 3.95336 × 10−26 | Plasma membrane |
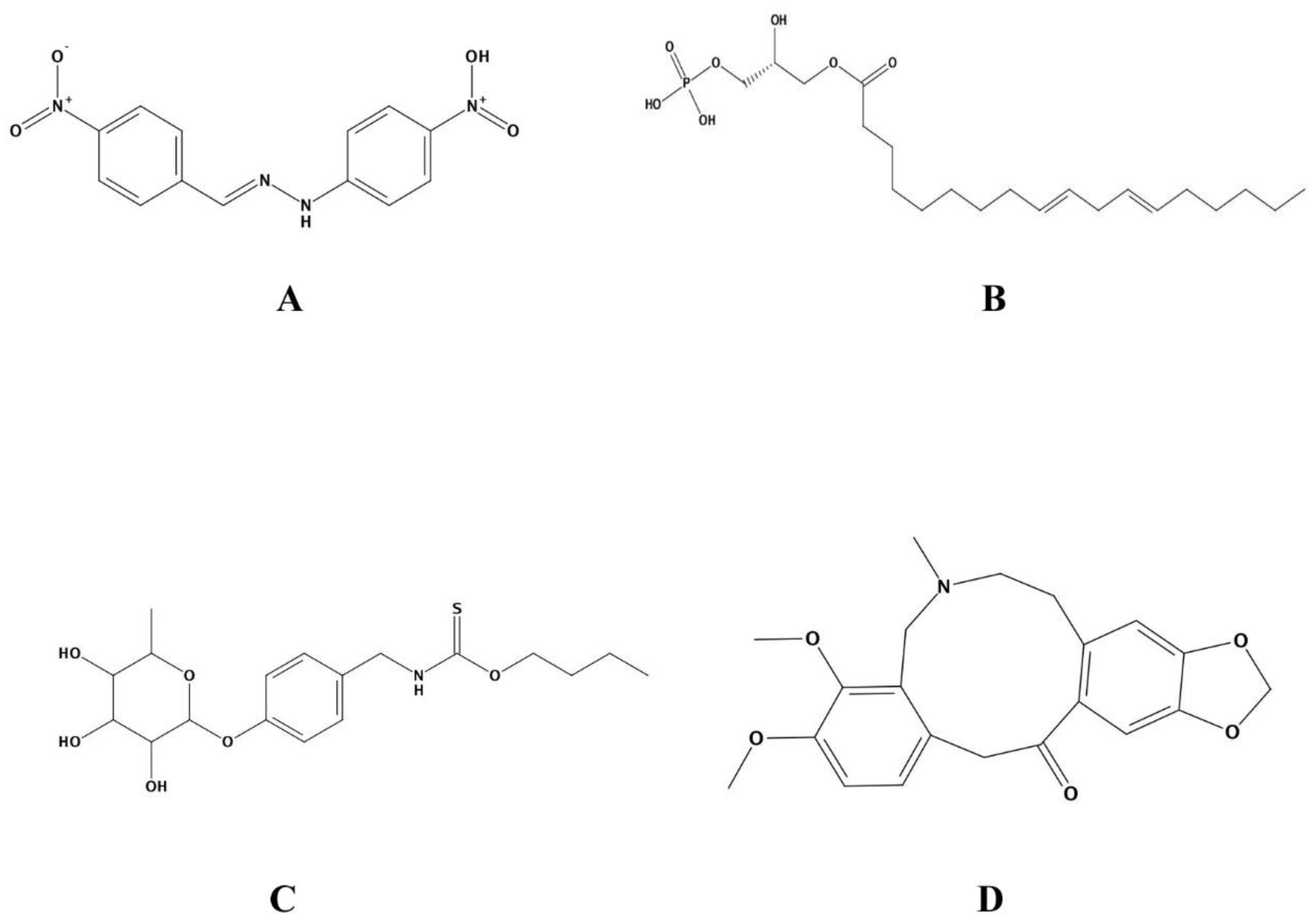
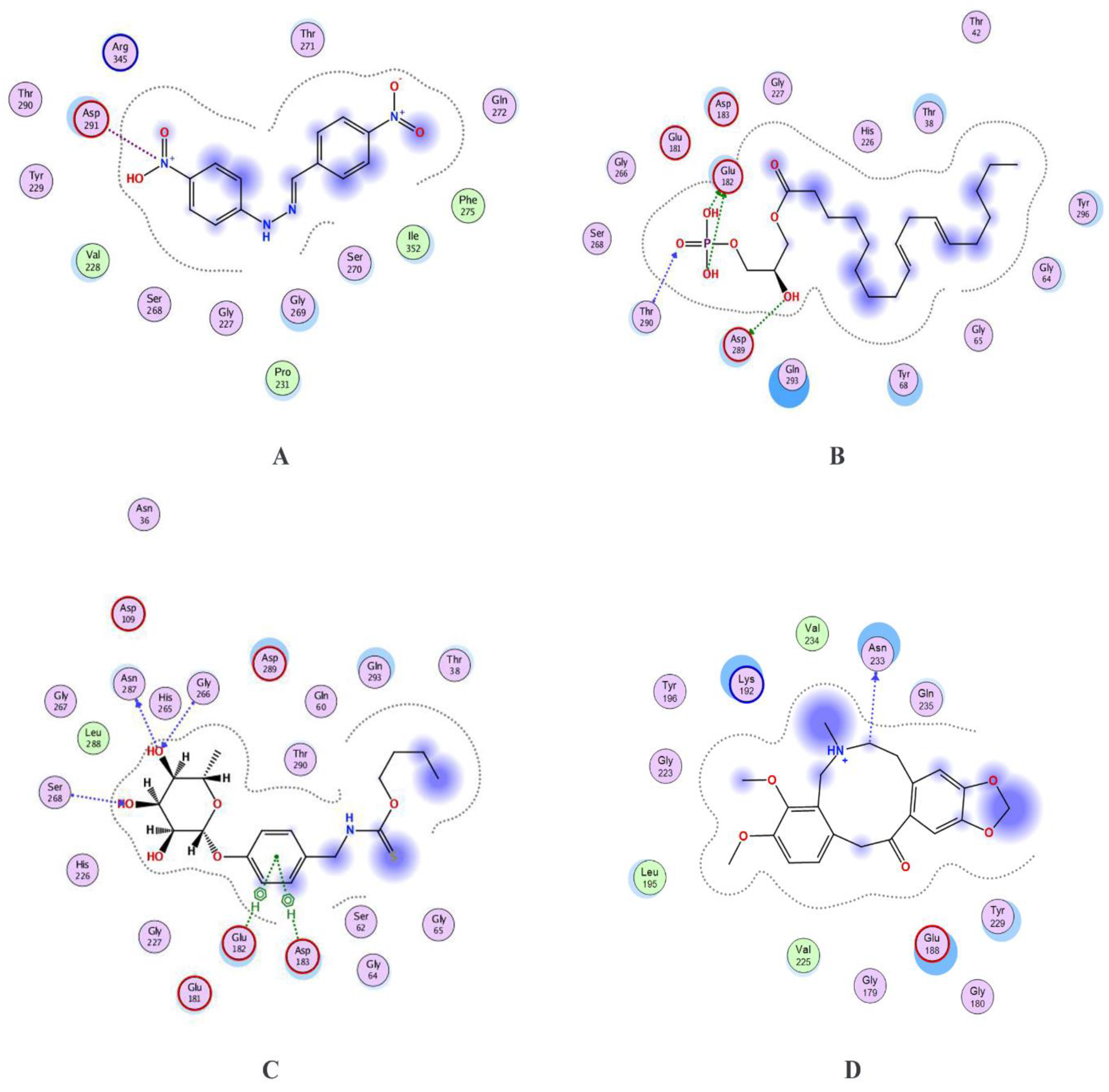
| Molecular Formula | Ligand Atom and Its Position | Receptor Atom/Residue | Interaction Type | Distance (Å) | Energy (Kcal/mol) | MM/PBSA Value of Complex | MM/PBSA Value of Ligand | |
|---|---|---|---|---|---|---|---|---|
| Control | C13H10N4O | N29 | OD1/ASP291 | ionic | 2.92 | −5.0 | −25.68 | 0.24 |
| ZINC13507461 | C21H39O7P | O57 | OD2/ASP289 | H-donor | 3.25 | −1.7 | −25.50 | −0.26 |
| O65 | OE2/GLU182 | H-donor | 2.99 | −5.3 | ||||
| O67 | OE1/GLU182 | H-donor | 2.87 | −7.1 | ||||
| O64 | N/THR290 | H-acceptor | 3.61 | −0.7 | ||||
| (4-Hydroxybenzyl)thiocarbamic acid | C8H9NO2S | O14 | O ASN287 | H-donor | 2.88 | −0.7 | −25.57 | −0.03 |
| O12 | N/SER268 | H-acceptor | 2.97 | −2.2 | ||||
| O12 | OG/SER268 | H-acceptor | 3.11 | −0.6 | ||||
| O14 | N/GLY266 | H-acceptor | 2.97 | −0.9 | ||||
| 6-ring | CA/GLU182 | pi-H | 3.92 | −0.7 | ||||
| 6-ring | N/ASP183 | pi-H | 4.61 | −0.9 | ||||
| Chelerythrine | C21H18NO4+ | C8 | O/ASN233 | H-donor | 3.38 | −0.6 | −25.64 | 0.11 |
| Property | Model Name | Unit | Predicted Value for Control | Predicted Value for ZINC13507461 | Predicted Value for (4-Hydroxybenzyl)thiocarbamic Acid | Predicted Value for Chelerythrine |
|---|---|---|---|---|---|---|
| Absorption | Water solubility | Numeric (log mol/L) | −3.655 | −4.445 | −2.833 | −3.123 |
| Caco2 permeability | Numeric (log Papp in 10−6 cm/s) | 0.222 | 0.521 | 0.41 | 1.429 | |
| Intestinal absorption (human) | Numeric (% absorbed) | 86.122 | 59.414 | 57.252 | 96.43 | |
| Skin permeability | Numeric (log Kp) | −2.766 | −2.702 | −3.041 | −2.946 | |
| P-glycoprotein substrate | Categorical (Yes/No) | Yes | Yes | Yes | No | |
| P-glycoprotein I inhibitor | Categorical (Yes/No) | No | Yes | No | Yes | |
| P-glycoprotein II inhibitor | Categorical (Yes/No) | No | Yes | No | Yes | |
| Distribution | VDss (human) | Numeric (log L/kg) | 0.531 | −0.866 | −0.716 | 0.53 |
| Fraction unbound (human) | Numeric (Fu) | 0.188 | 0.151 | 0.3 | 0.311 | |
| BBB permeability | Numeric (log BB) | −0.513 | −1.571 | −1.302 | 0.025 | |
| CNS permeability | Numeric (log PS) | −2.332 | −3.099 | −4.217 | −2.16 | |
| Metabolism | CYP3A4 substrate | Categorical (Yes/No) | No | Yes | No | Yes |
| CYP1A2 inhibitor | Categorical (Yes/No) | Yes | No | No | No | |
| CYP2C19 inhibitor | Categorical (Yes/No) | Yes | No | No | Yes | |
| CYP2D6 inhibitor | Categorical (Yes/No) | No | No | No | Yes | |
| Excretion | Total clearance | Numeric (log ml/min/kg) | 0.354 | 0.453 | 0.154 | 0.879 |
| Toxicity | Max. tolerated dose (human) | Numeric (log mg/kg/day) | 0.071 | 0.079 | 0.848 | 0.095 |
| Oral rat acute toxicity (LD50) | Numeric (mol/kg) | 2.513 | 2.985 | 3.023 | 3.411 | |
| Oral rat chronic toxicity (LOAEL) | Numeric (log mg/kg_bw/day) | 2.178 | 2.733 | 2.966 | 1.692 | |
| T. Pyriformis toxicity | Numeric (log ug/L) | 0.598 | 0.292 | 0.271 | 0.333 | |
| Minnow toxicity | Numeric (log mM) | 1.733 | −1.682 | 3.117 | 0.78 |
| Condition | Compounds | Intestinal Absorption of Compound Fa (%) | Portal Vein Absorption of Compound FDp (%) | Bioavailable Drug F (%) | Cmax (µg/mL) | Tmax (h) | AUC(0-inf) (ng-h/mL) | AUC(0-t) (ng-h/mL) |
|---|---|---|---|---|---|---|---|---|
| Healthy | Control | 81.626 | 80.078 | 25.145 | 3.3904 | 9.7593 | 1,034,000 | 27,820 |
| ZINC13507461 | 11.461 | 10.906 | 3.6584 | 0.4671 | 10 | 2622.2 | 2622.2 | |
| Chelerythrine | 99.582 | 99.42 | 31.551 | 4.3333 | 2.5876 | 226,700 | 38,260 | |
| (4-Hydroxybenzyl)thiocarbamic acid | 79.27 | 77.024 | 24.757 | 2.9855 | 8.8793 | 431,900 | 23,550 | |
| Cirrhosis | Control | 82.783 | 80.784 | 80.784 | 5.6819 | 9.8013 | 4,499,000 | 47,250 |
| ZINC13507461 | 11.565 | 11.025 | 11.025 | 1.0741 | 10 | 5728 | 5728 | |
| Chelerythrine | 99.903 | 99.866 | 99.866 | 0.8644 | 0.865 | 26,090,000 | 5852.2 | |
| (4-Hydroxybenzyl)thiocarbamic acid | 78.662 | 76.185 | 76.185 | 2.2192 | 9.9329 | 16,710 | 16,710 | |
| Renal impairment | Control | 82.792 | 80.896 | 80.896 | 4.9496 | 9.916 | 1,215,000 | 40,110 |
| ZINC13507461 | 11.694 | 11.148 | 11.148 | 1.0993 | 10 | 5819.1 | 5819.1 | |
| Chelerythrine | 99.645 | 99.487 | 31.45 | 4.2081 | 2.6187 | 225,600 | 37,180 | |
| (4-Hydroxybenzyl)thiocarbamic acid | 79.022 | 76.616 | 25.489 | 3.1081 | 8.8371 | 839,000 | 24,430 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basharat, Z.; Khan, K.; Jalal, K.; Alnasser, S.M.; Majeed, S.; Zehra, M. Inferring Therapeutic Targets in Candida albicans and Possible Inhibition through Natural Products: A Binding and Physiological Based Pharmacokinetics Snapshot. Life 2022, 12, 1743. https://doi.org/10.3390/life12111743
Basharat Z, Khan K, Jalal K, Alnasser SM, Majeed S, Zehra M. Inferring Therapeutic Targets in Candida albicans and Possible Inhibition through Natural Products: A Binding and Physiological Based Pharmacokinetics Snapshot. Life. 2022; 12(11):1743. https://doi.org/10.3390/life12111743
Chicago/Turabian StyleBasharat, Zarrin, Kanwal Khan, Khurshid Jalal, Sulaiman Mohammed Alnasser, Sania Majeed, and Marium Zehra. 2022. "Inferring Therapeutic Targets in Candida albicans and Possible Inhibition through Natural Products: A Binding and Physiological Based Pharmacokinetics Snapshot" Life 12, no. 11: 1743. https://doi.org/10.3390/life12111743
APA StyleBasharat, Z., Khan, K., Jalal, K., Alnasser, S. M., Majeed, S., & Zehra, M. (2022). Inferring Therapeutic Targets in Candida albicans and Possible Inhibition through Natural Products: A Binding and Physiological Based Pharmacokinetics Snapshot. Life, 12(11), 1743. https://doi.org/10.3390/life12111743