
Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme

 , , and
, , and 
Abstract
1. Introduction
2. Etiology of GBM
3. Neuropsychiatric Manifestations of GBM
4. Obesity: A Cause of Cancer
5. Understanding the General Causes of Obesity
5.1. A Sedentary Routine
5.2. Family History
5.3. Medicines
5.4. Smoking Cessation
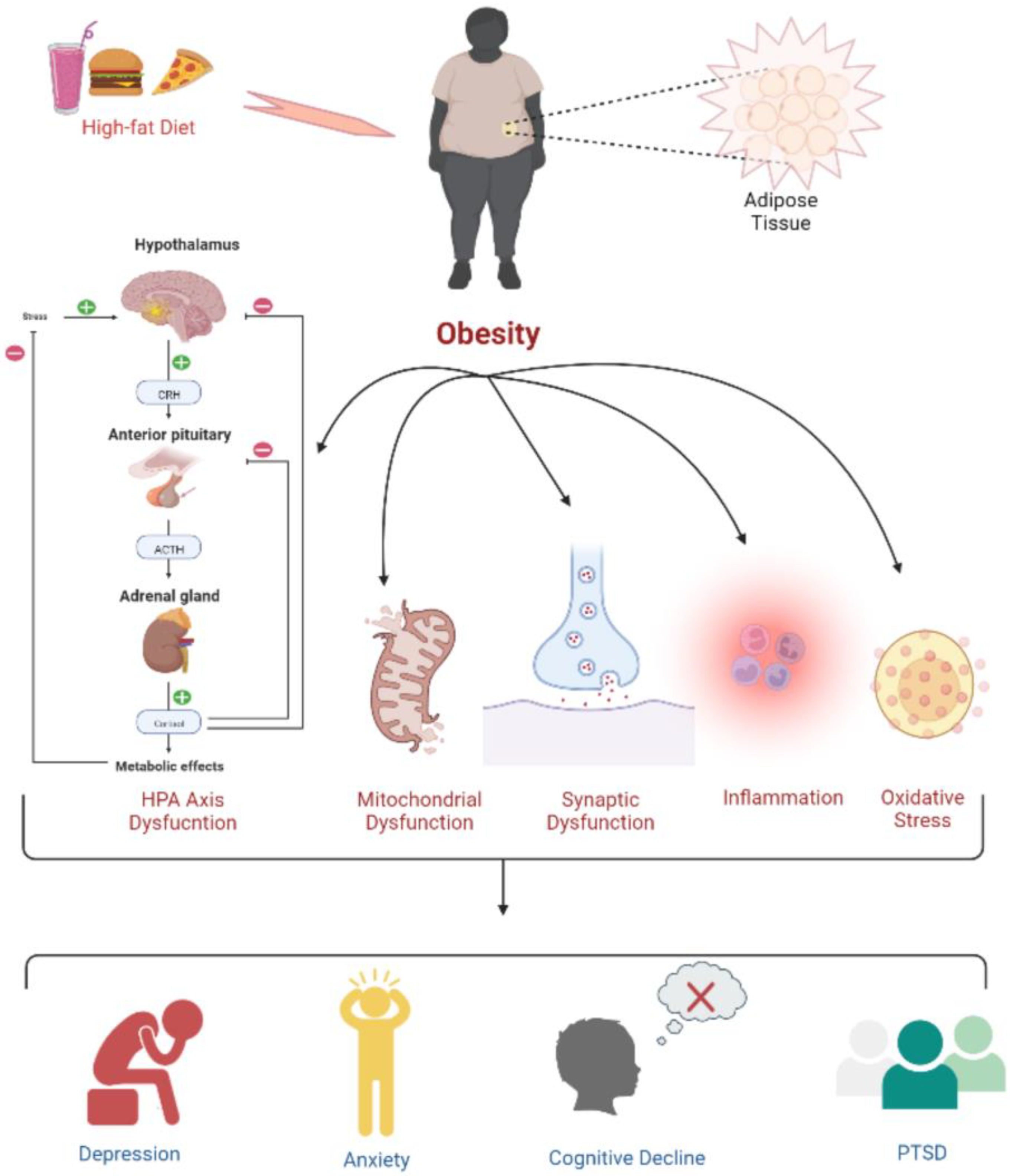
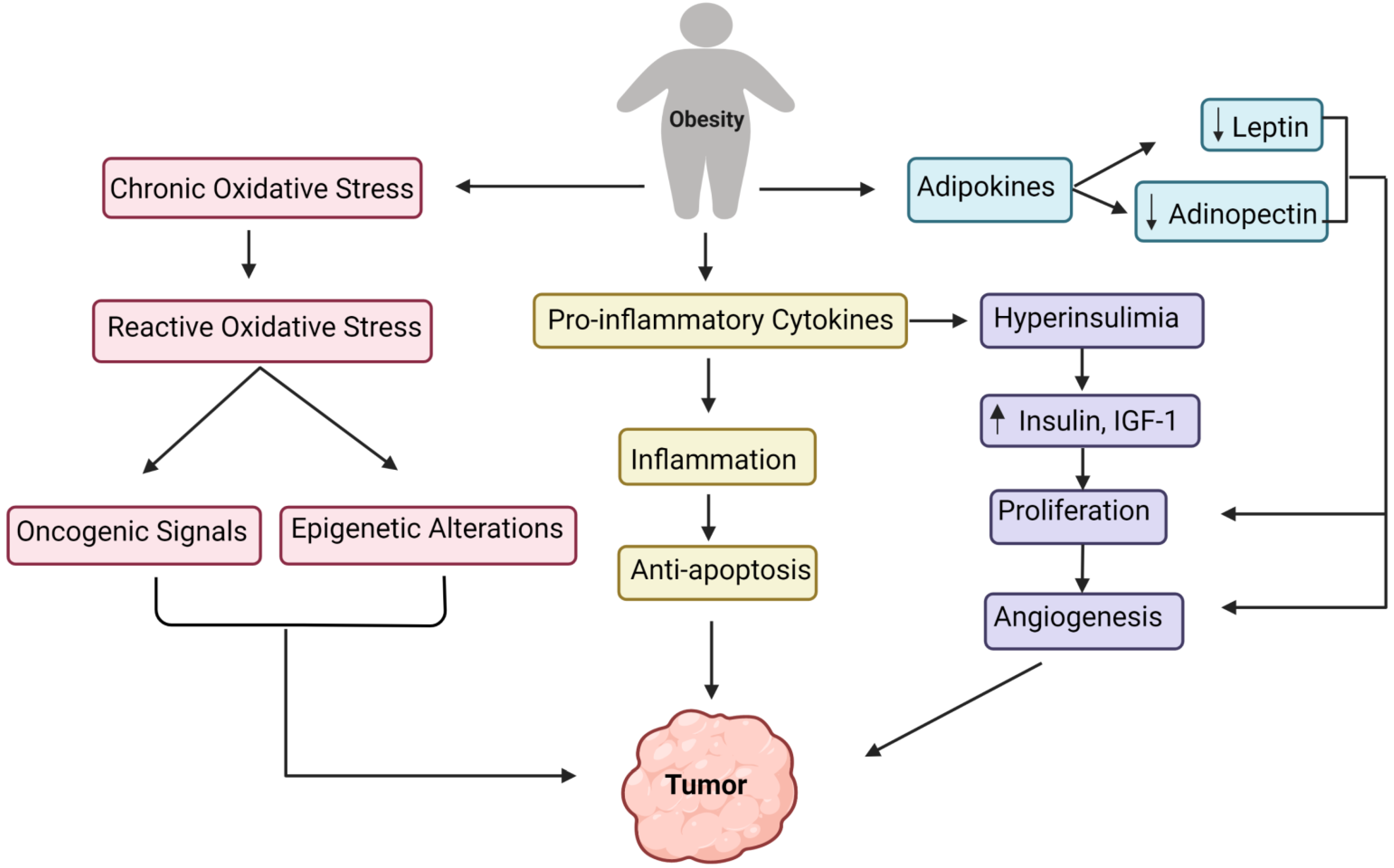
6. Development of Tumor Due to Obesity: Underlying the Biological Mechanism
7. Deciphering the Association of Obesity with the Development of GBM
8. Causes of Childhood Obesity: A Better Understanding of the Associated Concerns of Fatigue, Health Disorders, Cancer, and Death Risks in Children
8.1. Genetic Factors and Risk of Childhood Obesity
8.2. Endocrine Disorder and Childhood Obesity
8.2.1. Hypothyroidism
8.2.2. Cushing Syndrome or Hypercortisolism
8.2.3. Hypothalamic Obesity
8.2.4. Rapid Onset Obesity
8.3. Parent–Child Interactions: A Risk Factor for Childhood Obesity
9. Assessing the Risks of GBM with Childhood Obesity
10. Conclusions
Authors Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma multiforme |
| CNS | Central nervous system |
| BTs | Brain tumors |
| TCGA | The Cancer Genome Atlas |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| BMI | Body mass index |
| ASCVD | Atherosclerotic cardiovascular disease |
| SSRIs | Selective serotonin reuptake inhibitors |
| PTSD | Post-traumatic stress disorder |
| JNK | Jun N-terminal kinase |
| NFB | Nuclear factor B |
| MCP-1 | Monocyte chemoattractant protein-1 |
| TNF | Tumor necrosis factor |
| APN | Adiponectin |
| BMAds | Bone marrow adipocytes |
| RCC | Renal cell cancer |
| WC | Waist circumference |
| HL | Hodgkin lymphoma |
| HPA | Hypothalamic–pituitary–adrenal |
| MSCs | Mesenchymal stem cells |
| UCB | Umbilical cord blood |
| FTO | Fat mass and obesity-associated |
| m6A | N6-methyladenosine |
| IGF-1 | Insulin-like growth factor 1 |
| IGFIR | Insulin-like growth factor 1 receptor |
| LDs | Lipid droplets |
| TG | triglycerides |
| CE | Cholesteryl esters |
| ER | Endoplasmic reticulum |
| DIPG | Diffuse intrinsic pontine glioma |
| SCBT | Survivors of childhood brain tumors |
| MR | Mendelian randomization |
References
- DeAngelis, L.M.; Mellinghoff, I.K. Virchow 2011 or how to ID(H) human glioblastoma. J. Clin. Oncol. 2011, 29, 4473–4474. [Google Scholar] [PubMed]
- Ferguson, S.; Lesniak, M.S. Percival Bailey and the classification of brain tumors. Neurosurg. Focus. 2005, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.J. A critical review: The pathology of cerebral gliomas. J. Neurol. Psychiatry 1940, 3, 147–177. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.V.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar]
- Iacob, G.; Dinca, E.B. Current data and strategy in glioblastoma multiforme. J. Med. Life 2009, 2, 386–393. [Google Scholar]
- Soomro, S.H.; Ting, L.R.; Qing, Y.Y.; Ren, M. Molecular biology of glioblastoma: Classification and mutational locations. J. Pak. Med. Assoc. 2017, 67, 1410–1414. [Google Scholar]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Olar, A.; Aldape, K.D. Biomarkers classification and therapeutic decision-making for malignant gliomas. Curr. Treat. Options Oncol. 2012, 13, 417–436. [Google Scholar]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’Yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef]
- Ron, E.; Modan, B.; Boice, J.D., Jr.; Alfandary, E.; Stovall, M.; Chetrit, A.; Katz, L. Tumors of the brain and nervous system after radiotherapy in childhood. N. Engl. J. Med. 1988, 319, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Haas-Kogan, D.A. Radiation-induced gliomas. Expert Rev. Neurother. 2009, 9, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Simińska, D.; Korbecki, J.; Kojder, K.; Kapczuk, P.; Fabiańska, M.; Gutowska, I.; Machoy-Mokrzyńska, A.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Anthropometric Factors in Glioblastoma Multiforme-Literature Review. Brain Sci. 2021, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Gately, L.; McLachlan, S.A.; Dowling, A.; Philip, J. Life beyond a diagnosis of glioblastoma: A systematic review of the literature. J. Cancer Surviv. 2017, 11, 447–452. [Google Scholar] [CrossRef]
- Yamashita, S.; Saito, R.; Osawa, S.I.; Niizuma, K.; Ukishiro, K.; Kanamori, M.; Kakinuma, K.; Suzuki, K.; Tominaga, T. A Super-selective Wada Test Successfully Detected an Artery That Supplied Broca’s Area in a Case of Left Frontal Lobe Glioblastoma: Technical Case Report. Neurol. Med. Chir. 2021, 61, 661–666. [Google Scholar] [CrossRef]
- Habermeyer, B.; Weiland, M.; Mager, R.; Wiesbeck, G.A.; Wurst, F.M. A clinical lesson: Glioblastoma multiforme masquerading as depression in a chronic alcoholic. Alcohol Alcohol. 2008, 43, 31–33. [Google Scholar] [CrossRef]
- Gömöri, E.; Halbauer, J.D.; Kasza, G.; Varga, D.; Horvath, Z.; Komoly, S. Glioblastoma multiforme with an unusual location and clinical course. Clin. Neuropathol. 2009, 28, 165–167. [Google Scholar] [CrossRef]
- Mendez, G.; Ozpinar, A.; Raskin, J.; Gultekin, S.H.; Ross, D.A. Case comparison and literature review of glioblastoma: A tale of two tumors. Surg. Neurol. Int. 2014, 5, 121. [Google Scholar]
- Schwab, R.; Kubik, C.S. Glioblastoma multiforme of left temporal lobe, with metastases to spinal cord and spinal nerves. N. Engl. J. Med. 1949, 241, 939. [Google Scholar] [PubMed]
- Dietch, J.T. Cerebral tumor presenting with panic attacks. Psychosomatics 1984, 25, 861–863. [Google Scholar] [CrossRef]
- Yoshikawa, A.; Nakada, M.; Watanabe, T.; Hayashi, Y.; Sabit, H.; Kato, Y.; Suzuki, S.; Ooi, A.; Sato, H.; Hamada, J.-I. Progressive adult primary glioblastoma in the medulla oblongata with an unmethylated MGMT promoter and without an IDH mutation. Brain Tumor. Pathol. 2013, 30, 175–179. [Google Scholar] [CrossRef]
- Moise, D.; Madhusoodanan, S. Psychiatric symptoms associated with brain tumors: A clinical enigma. CNS Spectr. 2006, 11, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sinha, J.K.; Raghunath, M. ‘Obesageing’: Linking obesity & ageing. Indian J. Med. Res. 2019, 149, 610–615. [Google Scholar]
- Jovanov, E.; Sazonov, E.; Poon, C. Sensors and systems for obesity care and research. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; pp. 3188–3191. [Google Scholar]
- Calle, E.E.; Thun, M.J. Obesity and cancer. Oncogene 2004, 23, 6365–6378. [Google Scholar] [CrossRef]
- Amer, A.; Franchi, L.; Kanneganti, T.D.; Body-Malapel, M.; Özören, N.; Brady, G.; Meshinchi, S.; Jagirdar, R.; Gewirtz, A.; Akira, S.; et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 2006, 281, 35217–35223. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef]
- Ferguson, R.D.; Novosyadlyy, R.; Fierz, Y.; Alikhani, N.; Sun, H.; Yakar, S.; LeRoith, D. Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res. 2012, 14, R8. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef]
- Olivas, A.; Price, R.S. Obesity, Inflammation, and Advanced Prostate Cancer. Nutr. Cancer 2021, 73, 2232–2248. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Obesity-associated Breast Cancer: Analysis of risk factors. Adv. Exp. Med. Biol. 2017, 960, 571–606. [Google Scholar]
- Hidayat, K.; Du, X.; Chen, G.; Shi, M.; Shi, B. Abdominal Obesity and Lung Cancer Risk: Systematic Review and Meta-Analysis of Prospective Studies. Nutrients 2016, 8, 810. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, J.L.; Liss, M.A.; Parsons, J.K. Obesity, Physical Activity and Bladder Cancer. Curr. Urol. Rep. 2015, 16, 74. [Google Scholar] [CrossRef]
- Bardou, M.; Barkun, A.N.; Martel, M. Obesity and colorectal cancer. Gut 2013, 62, 933–947. [Google Scholar] [CrossRef]
- Wilson, K.M.; Cho, E. Obesity and Kidney Cancer. Recent Results Cancer Res. 2016, 208, 81–93. [Google Scholar]
- Matos, A.L.; arinho-Dias, J.; Ramalheira, S.; Oliveira, M.J.; Bicho, M.; Ribeiro, R. Mechanisms underlying the association between obesity and Hodgkin lymphoma. Tumour. Biol. 2016, 37, 13005–13016. [Google Scholar] [CrossRef]
- Olszańska, J.; Pietraszek-Gremplewicz, K.; Nowak, D. Melanoma Progression under Obesity: Focus on Adipokines. Cancers 2021, 13, 2281. [Google Scholar] [CrossRef]
- Rawla, P.; Thandra, K.C.; Sunkara, T. Pancreatic cancer and obesity: Epidemiology, mechanism, and preventive strategies. Clin. J. Gastroenterol. 2019, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, Y.; Zhou, T.; Li, B.; Wang, Z. Adiponectin and Thyroid Cancer: Insight into the Association between Adiponectin and Obesity. Aging Dis. 2021, 12, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Ray, I.; Meira, L.B.; Michael, A.; Ellis, P.E. Adipocytokines and disease progression in endometrial cancer: A systematic review. Cancer Metastasis Rev. 2022, 41, 211–242. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; Fairfield, H.; Rosen, C.J. Bone Marrow Adipocytes: A Link between Obesity and Bone Cancer. Cancers 2021, 13, 364. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, J.; Zhou, Y.; Qiao, L. Obesity and gastric cancer. Front. Biosci. 2012, 17, 2383–2390. [Google Scholar] [CrossRef]
- Lim, S.H.; Jeon, S.Y.; Jeon, I.S.; Kang, M.H. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation associated with neuroblastoma. Pediatr. Blood Cancer 2018, 65, e26983. [Google Scholar] [CrossRef]
- Wright, S.M.; Aronne, L.J. Causes of obesity. Abdom. Imaging 2012, 37, 730–732. [Google Scholar] [CrossRef]
- Wadden, T.A.; Tronieri, J.S.; Butryn, M.L. Lifestyle modification approaches for the treatment of obesity in adults. Am. Psychol. 2020, 75, 235–251. [Google Scholar] [CrossRef]
- Romero-Ibarguengoitia, M.E.; Vadillo-Ortega, F.; Caballero, A.E.; Ibarra-González, I.; Herrera-Rosas, A.; Serratos-Canales, M.F.; León-Hernández, M.; González-Chávez, A.; Mummidi, S.; Duggirala, R.; et al. Family history and obesity in youth, their effect on acylcarnitine/minoacids metabolomics and non-alcoholic fatty liver disease (NAFLD). Structural equation modeling approach. PLoS ONE 2018, 13, e0193138. [Google Scholar]
- Sakata, T.; Yoshimatsu, H.; Kurokawa, M. Hypothalamic neuronal histamine: Implications of its homeostatic control of energy metabolism. Nutrition 1997, 13, 403–411. [Google Scholar] [CrossRef]
- Ness-Abramof, R.; Apovian, C.M. Drug-induced weight gain. Drugs Today 2005, 41, 547–555. [Google Scholar] [CrossRef]
- Chukwu, J.; Delanty, N.; Webb, D.; Cavalleri, G.L. Weight change, genetics and antiepileptic drugs. Expert Rev. Clin. Pharmacol. 2014, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.C.; Alloway, R.R.; Buell, J.F.; Boardman, R.; Alexander, J.W.; Cardi, M.; Roy-Chaudhury, P.; First, M.R.; Succop, P.; Munda, R.; et al. Body weight alterations under early corticosteroid withdrawal and chronic corticosteroid therapy with modern immunosuppression. Transplantation 2005, 80, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Clineschmidt, B.V.; Lotti, V.J. Histamine: Intraventricular injection suppresses ingestive behavior of the cat. Arch. Int. Pharmacodyn. Ther. 1973, 206, 288–298. [Google Scholar] [PubMed]
- Parsons, M.E.; Ganellin, C.R. Histamine and its receptors. Br. J. Pharmacol. 2006, 147, S127–S135. [Google Scholar] [CrossRef]
- Aubin, H.J.; Farley, A.; Lycett, D.; Lahmek, P.; Aveyard, P. Weight gain in smokers after quitting cigarettes: Meta-analysis. BMJ 2012, 345, 4439. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D. Pathophysiology of spinal cord injury. Current and future therapies. Minerva Anestesiol. 1989, 55, 63–66. [Google Scholar]
- Hur, Y.N.; Hong, G.H.; Choi, S.H.; Shin, K.H.; Chun, B.G. High fat diet altered the mechanism of energy homeostasis induced by nicotine and withdrawal in C57BL/6 mice. Mol. Cells 2010, 30, 219–226. [Google Scholar] [CrossRef]
- Lerman, C.; Berrettini, W.; Pinto, A.; Patterson, F.; Crystal-Mansour, S.; Wileyto, E.P.; Restine, S.L.; Leonard, D.G.; Shields, P.G.; Epstein, L.H. Changes in food reward following smoking cessation: A pharmacogenetic investigation. Psychopharmacology 2004, 174, 571–577. [Google Scholar] [CrossRef]
- Harris, K.K.; Zopey, M.; Friedman, T.C. Metabolic effects of smoking cessation. Nat. Rev. Endocrinol. 2016, 12, 684. [Google Scholar] [CrossRef]
- White, M.A.; Masheb, R.M.; Grilo, C.M. Self-reported weight gain following smoking cessation: A function of binge eating behavior. Int. J. Eat. Disord. 2010, 43, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Telang, F. Overlapping neuronal circuits in addiction and obesity: Evidence of systems pathology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.M.; Hollander, J.A.; Kenny, P.J. Decreased brain reward function during nicotine withdrawal in C57BL6 mice: Evidence from intracranial self-stimulation (ICSS) studies. Pharmacol. Biochem. Behav. 2008, 90, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 2004, 89, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, A.H. Obesity: A modern epidemic. Trans. Am. Clin. Climatol. Assoc. 2005, 116, 103–113. [Google Scholar] [PubMed]
- Bifulco, M.; Pisanti, S. “Adiponcosis”: A new term to name the obesity and cancer link. J. Clin. Endocrinol. Metab. 2013, 98, 4664–4665. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 02078. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Akıncılar, S.C.; Tergaonkar, V. Chronic adipose tissue inflammation: All immune cells on the stage. Trends Mol. Med. 2013, 19, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Hodson, L. The influence of dietary fat on liver fat accumulation. Nutrients 2014, 6, 5018–5033. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, J.; Cereijo, R.; Gavaldà-Navarro, A.; Peyrou, M.; Giralt, M.; Villarroya, F. New insights into the secretory functions of brown adipose tissue. J. Endocrinol. 2019, 243, R19–R27. [Google Scholar] [CrossRef]
- Akimoto, K.; Kimura, K.; Nagano, M.; Takano, S.; Salazar, G.; Yamashita, T.; Ohneda, O. Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem. Cells Dev. 2013, 22, 1370–1386. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Du, B. Novel positioning from obesity to cancer: FTO, an m6A RNA demethylase, regulates tumour progression. J. Cancer Res. Clin. Oncol. 2019, 145, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, Insulin, and Cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef]
- El-Roeiy, A.; Chen, X.; Roberts, V.J.; Shimasakai, S.; Ling, N.; Leroith, D.; Roberts, C.T.; Yen, S.S. Expression of the genes encoding the insulin-like growth factors (IGF-I and II), the IGF and insulin receptors, and IGF-binding proteins-1-6 and the localization of their gene products in normal and polycystic ovary syndrome ovaries. J. Clin. Endocrinol. Metab. 1994, 78, 1488–1496. [Google Scholar]
- Gascoigne, A.D.; Richards, J.; Gould, K.; Gibson, G.J. Successful treatment of Bacillus cereus infection with ciprofloxacin. Thorax 1991, 46, 220–221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vishwamitra, D.; George, S.K.; Shi, P.; Kaseb, A.O.; Amin, H.M. Type I insulin-like growth factor receptor signaling in hematological malignancies. Oncotarget 2017, 8, 1814–1844. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C.; Bryson, J.M.; Turtle, J.R. Somatogenic receptors of rat liver: Regulation by insulin. Endocrinology 1980, 107, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, S.; Tian, T.; Bai, J.; Hu, Z.; Xu, Y.; Dong, J.; Chen, F.; Wang, X.; Shen, H. Phenotypes and genotypes of insulin-like growth factor 1, IGF-binding protein-3 and cancer risk: Evidence from 96 studies. Eur. J. Hum. Genet. 2009, 17, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; van Golen, C.M.; Feldman, E.L. Insulin-like growth factor-I signaling in human neuroblastoma cells. Oncogene 2004, 23, 130–141. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.Z.; Wong, Y.C. Sex hormone-induced prostatic carcinogenesis in the noble rat: The role of insulin-like growth factor-I (IGF-I) and vascular endothelial growth factor (VEGF) in the development of prostate cancer. Prostate 1998, 35, 165–177. [Google Scholar] [CrossRef]
- Poloz, Y.; Stambolic, V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 2015, 6, e2037. [Google Scholar] [CrossRef]
- Dupont, J.; LeRoith, D. Insulin and insulin-like growth factor I receptors: Similarities and differences in signal transduction. Horm. Res. 2001, 55 (Suppl. 2), 22–26. [Google Scholar] [CrossRef]
- Liu, T.J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef]
- Gong, Y.; Ma, Y.; Sinyuk, M.; Loganathan, S.; Thompson, R.C.; Sarkaria, J.N.; Chen, W.; Lathia, J.D.; Mobley, B.C.; Clark, S.W.; et al. Insulin-mediated signaling promotes proliferation and survival of glioblastoma through Akt activation. Neuro. Oncol. 2016, 18, 48–57. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. The life of lipid droplets. Biochim. Biophys. Acta 2009, 1791, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Beckman, M. Cell biology. Great balls of fat. Science 2006, 311, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3, 1–11. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Mohammed, M.S.; Sendra, S.; Lloret, J.; Bosch, I. Systems and WBANs for Controlling Obesity. J. Healthc. Eng. 2018, 2018, 1564748. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, T. Childhood obesity. Panminerva. Med. 2018, 60, 200–212. [Google Scholar] [CrossRef]
- Bouchard, C. Genetic determinants of regional fat distribution. Hum. Reprod. 1997, 12 (Suppl. 1), 1–5. [Google Scholar] [CrossRef]
- Dubern, B.; Bisbis, S.; Talbaoui, H.; Le Beyec, J.; Tounian, P.; Lacorte, J.-M.; Clément, K. Homozygous null mutation of the melanocortin-4 receptor and severe early-onset obesity. J. Pediatr. 2007, 150, 613–617.e1. [Google Scholar] [CrossRef]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Neu, J. Early factors leading to later obesity: Interactions of the microbiome, epigenome, and nutrition. Curr. Probl. Pediatr. Adolesc. Health Care 2015, 45, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Jebeile, H.; Kelly, A.S.; O’Malley, G.; Baur, L.A. Obesity in children and adolescents: Epidemiology, causes, assessment, and management. Lancet Diabetes Endocrinol. 2022, 10, 351–365. [Google Scholar] [CrossRef]
- Wagner, E.; Jamil, O.; Hodges, B. Talking About Childhood Obesity. Clin. Pediatr. 2022, 61, 266–269. [Google Scholar] [CrossRef]
- Verma, N.; Jain, V. Iatrogenic Cushing syndrome. Indian Pediatr. 2012, 49, 765. [Google Scholar] [CrossRef][Green Version]
- Crocker, M.K.; Yanovski, J.A. Pediatric obesity: Etiology and treatment. Pediatr. Clin. N. Am. 2011, 58, 1217–1240. [Google Scholar] [CrossRef]
- Bougnères, P.; Pantalone, L.; Linglart, A.; Rothenbühler, A.; Le Stunff, C. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J. Clin. Endocrinol. Metab. 2008, 93, 3971–3980. [Google Scholar] [CrossRef]
- Anderson, S.E.; Keim, S.A. Parent-Child Interaction, Self-Regulation, and Obesity Prevention in Early Childhood. Curr. Obes. Rep. 2016, 5, 192–200. [Google Scholar] [CrossRef]
- Schore, A.N. Back to basics: Attachment, affect regulation, and the developing right brain: Linking developmental neuroscience to pediatrics. Pediatr. Rev. 2005, 26, 204–217. [Google Scholar] [CrossRef]
- McEwen, B.S. Understanding the potency of stressful early life experiences on brain and body function. Metabolism 2008, 57 (Suppl. 2), S11–S15. [Google Scholar] [CrossRef]
- Peters, A.; Kubera, B.; Hubold, C.; Langemann, D. The selfish brain: Stress and eating behavior. Front. Neurosci. 2011, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Langemann, D. Stress and eating behavior. F1000 Biol. Rep. 2010, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Papies, E.K.; Stroebe, W.; Aarts, H. Healthy cognition: Processes of self-regulatory success in restrained eating. Personal. Soc. Psychol. Bull. 2008, 34, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.A.; Susman, E.J. Self-regulation and rapid weight gain in children from age 3 to 12 years. Arch. Pediatr. Adolesc. Med. 2009, 163, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.S.; Bradley, L.J.; Brolin, R.E. Gastric bypass surgery in adolescents with morbid obesity. J. Pediatr. 2001, 138, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.S.; Barlow, S.E.; Rao, G.; Inge, T.H.; Hayman, L.L.; Steinberger, J.; Urbina, E.M.; Ewing, L.J.; Daniels, S.R.; on behalf of the American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young, Council on Nutrition, Physical Activity and Metabolism, and Council on Clinical Cardiology. Severe obesity in children and adolescents: Identification, associated health risks, and treatment approaches: A scientific statement from the American Heart Association. Circulation 2013, 128, 1689–1712. [Google Scholar]
- Eagle, T.F.; Sheetz, A.; Gurm, R.; Woodward, A.C.; Kline-Rogers, E.; Leibowitz, R.; Durussel-Weston, J.; Palma-Davis, L.; Aaronson, S.; Fitzgerald, C.M.; et al. Understanding childhood obesity in America: Linkages between household income, community resources, and children’s behaviors. Am. Heart J. 2012, 163, 836–843. [Google Scholar] [CrossRef]
- Pineda, E.; Sanchez-Romero, L.M.; Brown, M.; Jaccard, A.; Jewell, J.; Galea, G.; Webber, L.; Breda, J. Forecasting Future Trends in Obesity across Europe: The Value of Improving Surveillance. Obes. Facts 2018, 11, 360–371. [Google Scholar] [CrossRef]
- Ghosh, S.; Sachdeva, B.; Sachdeva, P.; Chaudhary, V.; Rani, G.M.; Sinha, J.K. Graphene quantum dots as a potential diagnostic and therapeutic tool for the management of Alzheimer’s disease. Carbon. Lett. 2022. [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro. Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Wilne, S.; Collier, J.; Kennedy, C.; Koller, K.; Grundy, R.; Walker, D. Presentation of childhood CNS tumours: A systematic review and meta-analysis. Lancet Oncol. 2007, 8, 685–695. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J. Neurooncol. 2017, 134, 541–549. [Google Scholar] [CrossRef]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Qaddoumi, I.; Kocak, M.; Panandiker, P.A.S.; Armstrong, G.T.; Wetmore, C.; Crawford, J.R.; Lin, T.; Boyett, J.M.; Kun, L.E.; Boop, F.A.; et al. Phase II Trial of Erlotinib during and after Radiotherapy in Children with Newly Diagnosed High-Grade Gliomas. Front. Oncol. 2014, 4, 67. [Google Scholar] [CrossRef]
- Papalia, H.; Rochette, E.; Pereira, B.; Merlin, E.; Kanold, J.; Duché, P. Metabolic response to exercise in childhood brain tumor survivors: A pilot controlled study. Pediatr. Blood Cancer 2020, 67, e28053. [Google Scholar] [CrossRef]
- Howell, A.E.; Robinson, J.W.; Wootton, R.E.; McAleenan, A.; Tsavachidis, S.; Ostrom, Q.T.; Bondy, M.; Armstrong, G.; Relton, C.; Haycock, P.; et al. Testing for causality between systematically identified risk factors and glioma: A Mendelian randomization study. BMC Cancer 2020, 20, 508. [Google Scholar] [CrossRef]
- Green, D.M.; Cox, C.L.; Zhu, L.; Krull, K.R.; Srivastava, D.K.; Stovall, M.; Nolan, V.G.; Ness, K.K.; Donaldson, S.S.; Oeffinger, K.C.; et al. Risk factors for obesity in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2012, 30, 246–255. [Google Scholar] [CrossRef]
- Thompson, E.M.; Donnai, D.; Baraitser, M.; Hall, C.M.; Pembrey, M.E.; Fixsen, J. Multiple pterygium syndrome: Evolution of the phenotype. J. Med. Genet. 1987, 24, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Bufalieri, F.; Basili, I.; Di Marcotullio, L.; Infante, P. Harnessing the Activation of RIG-I Like Receptors to Inhibit Glioblastoma Tumorigenesis. Front. Mol. Neurosci. 2021, 14, 710171. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Meng, M.; Cao, C.; Zhang, J.; Cheng, X.; Huang, Y.; Cao, H.; Li, Y.; Tian, D.; Huang, Y.; et al. RNA-binding protein MEX3A controls G1/S transition via regulating the RB/E2F pathway in clear cell renal cell carcinoma. Mol. Ther. Nucleic. Acids. 2021, 27, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Disney-Hogg, L.; Sud, A.; Law, P.J.; Cornish, A.J.; Kinnersley, B.; Ostrom, Q.T.; Labreche, K.; Eckel-Passow, J.E.; Armstrong, G.N.; Claus, E.B.; et al. Influence of obesity-related risk factors in the aetiology of glioma. Br. J. Cancer 2018, 118, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Sinha, J.K.; Ghosh, S.; Swain, U.; Giridharan, N.V.; Raghunath, M. Increased macromolecular damage due to oxidative stress in the neocortex and hippocampus of WNIN/Ob, a novel rat model of premature aging. Neuroscience 2014, 269, 256–264. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Raghunath, M. Epigenomic maintenance through dietary intervention can facilitate DNA repair process to slow down the progress of premature aging. IUBMB Life 2016, 68, 717–721. [Google Scholar] [CrossRef]
- Sinha, J.K.; Vashisth, K.; Ghosh, S. The importance of sleep studies in improving the health indices of a nation. Sleep Med. X 2022, 4, 100049. [Google Scholar] [CrossRef]
- Ghosh, S.; Raghunath, M.; Das, B.C.; Sinha, J.K. High sugar content in baby food: An Indian perspective. Lancet Diabetes Endocrinol. 2019, 7, 748–749. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| S. No. | Patient’s Age | Patient’s Gender | Location of GBM Tumor | Neuropsychiatric Symptoms | Reference |
|---|---|---|---|---|---|
| 1. | 37 years | Male | Left frontal lobe | Headache, anomic aphasia, paraphasia, and dysgraphia | [17] |
| 2. | 34 years | Male | Right frontal lobe | Delirium, depression | [18] |
| 3. | 39 years | Male | Frontal bilateral lobes | Aphasia, headache, anxiety, PTSD, left side numbness | [19] |
| 4. | 63 years | Male | Right temporal lobe | Partial complex seizure | [20] |
| 5. | 59 years | Male | Left temporal lobe | Slurred speech, memory impairment, clonic movement of arms and legs, weakness, sharp pain in limbs, and rapid clonic tremor | [21] |
| 6. | 55 years | Female | Left frontoparietal lobes | Panic attacks, agoraphobia. Depression and Right-sided weakness | [22] |
| 7. | 63 years | Female | Medulla oblongata | Gait disturbances, dizziness, loss of appetite | [23] |
| 8. | 29 years | Female | Left thalamus | Memory impairment, borderline personality disorder, depressive symptoms, PTSD | [24] |
| S. No. | Cancer Type | Association with Obesity | Reference |
|---|---|---|---|
| 1. | Prostate Cancer | Obesity promotes low-grade inflammation linked to prostate cancer progression by compromising treatment and diagnosis. | [33] |
| 2. | Breast Cancer | In obese women, aromatization activity due to elevated levels of estrogen, the overexpression of insulin resistance, proinflammatory cytokines, IGFs, oxidative stress, and hypercholesterolemia contributes to the development of breast cancer. | [34] |
| 3. | Lung Cancer | Abdominal obesity has a significant role in the development of lung cancer. Smoking is one of the leading causes of the development of lung cancer; people who smoke regularly have an increased BMI. The reduced levels of sex-hormone-binding globulin and elevated estrogens and androgens are associated with obesity and an increased risk of lung cancer. However, the exact mechanism is not understood. | [35] |
| 4. | Bladder cancer | Obesity is a potential risk factor for recurrence, progression, or death with bladder cancer. | [36] |
| 5. | Colorectal Cancer | Visceral and abdominal fat increase the risks of colorectal cancer by up to 30–70% in men and are linked with worse outcomes and recurrence. | [37] |
| 6. | Kidney Cancer | Renal cell cancer (RCC) is the main form of kidney cancer. Studies have reported that people with a high waist-to-hip ratio (WHR), waist circumference (WC), and increased BMI are risk factors associated with RCC. | [38] |
| 7. | Hodgkin Lymphoma (HL) | It has been elucidated that inflammation is common both in HL and obesity. The interaction of molecules released by adipocytes and the tumor microenvironment associates obesity with an increased risk of developing HL. | [39] |
| 8. | Melanoma | Adipocytes provide nutrients to melanoma cells. Adipokines released by adipocytes stimulate the progression of myeloma cells. Moreover, it has been reported that insulin resistance and hyperinsulinemia may promote the growth of myeloma. | [40] |
| 9. | Pancreatic Cancer | Obesity increases the risk of pancreatic cancer through mechanisms that are not fully understood. Inflammation and hormone imbalance could be plausible causes. Excess abdominal adiposity is one of the few controllable risk factors for developing pancreatic cancer. | [41] |
| 10. | Thyroid Cancer | Adiponectin (APN) is one of the vitally essential adipocytokines. In obese individuals, there are reduced levels of APN. Similarly, reduced levels of APN have been found in patients with thyroid cancer and metabolic syndrome. | [42] |
| 11. | Endometrial Cancer | Visfatin, leptin, and resistin are associated with endometrial cancer proliferation, growth metastasis, and invasion. | [43] |
| 12. | Bone Cancer | Bone marrow adipocytes (BMAds) increase in size and number during obesity and can initiate bone cancer or cancer within the bone marrow. The BMAds provide nutrients to tumor cells and help in tumor cell proliferation. | [44] |
| 13. | Gastric Cancer | Obesity is associated with the occurrence of gastric cancer. The mechanism involved includes insulin resistance; higher levels of IGFs; and altered leptin, ghrelin, and adiponectin levels. | [45] |
| 14. | Neuroblastoma | Lim et al. reported on a 29-month-old Korean female who developed neuroblastoma and showed clinical features of ROHHAD. The laboratory examinations revealed high levels of IGF-1, prolactin, sex hormone, cortisol, and lactate dehydrogenase. | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sachdeva, P.; Ghosh, S.; Ghosh, S.; Han, S.; Banerjee, J.; Bhaskar, R.; Sinha, J.K. Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme. Life 2022, 12, 1673. https://doi.org/10.3390/life12101673
Sachdeva P, Ghosh S, Ghosh S, Han S, Banerjee J, Bhaskar R, Sinha JK. Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme. Life. 2022; 12(10):1673. https://doi.org/10.3390/life12101673
Chicago/Turabian StyleSachdeva, Punya, Shampa Ghosh, Soumya Ghosh, Sungsoo Han, Juni Banerjee, Rakesh Bhaskar, and Jitendra Kumar Sinha. 2022. "Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme" Life 12, no. 10: 1673. https://doi.org/10.3390/life12101673
APA StyleSachdeva, P., Ghosh, S., Ghosh, S., Han, S., Banerjee, J., Bhaskar, R., & Sinha, J. K. (2022). Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme. Life, 12(10), 1673. https://doi.org/10.3390/life12101673