
Whole Exome-Sequencing of Pooled Genomic DNA Samples to Detect Quantitative Trait Loci in Esotropia and Exotropia of Strabismus in Japanese


Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. DNA Sequence
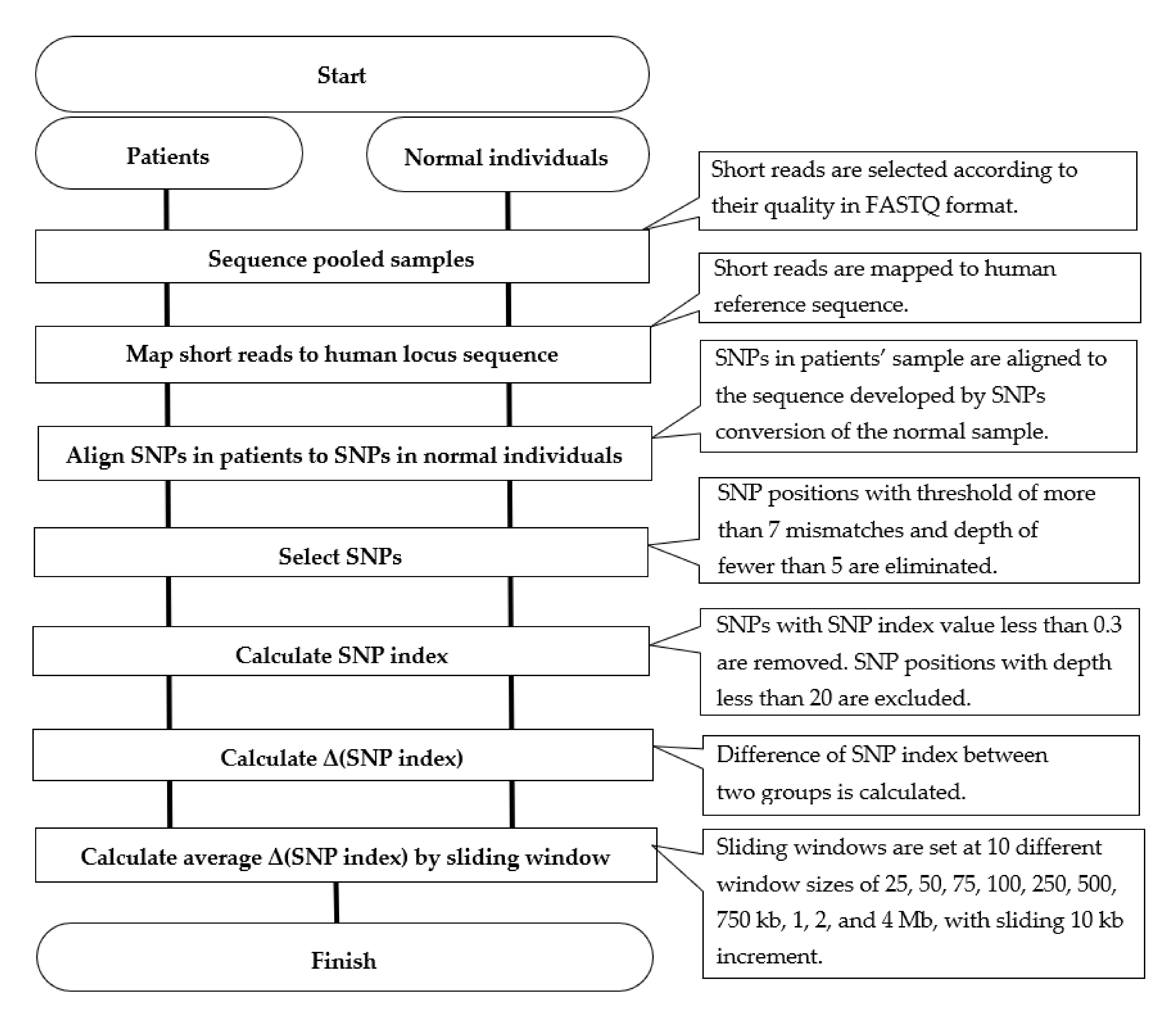
2.3. Sequencing Data Analysis and Generation of SNP Index
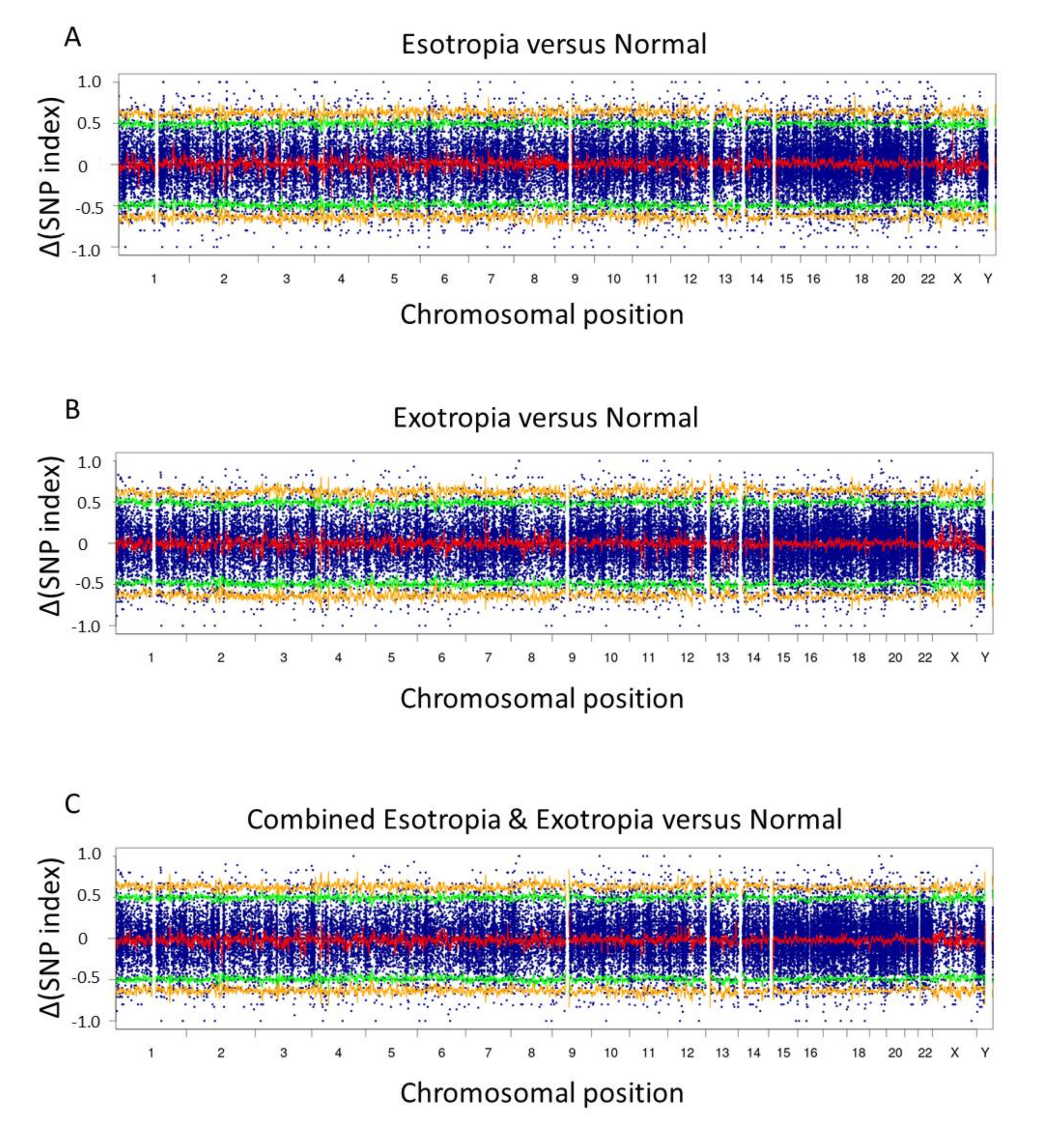
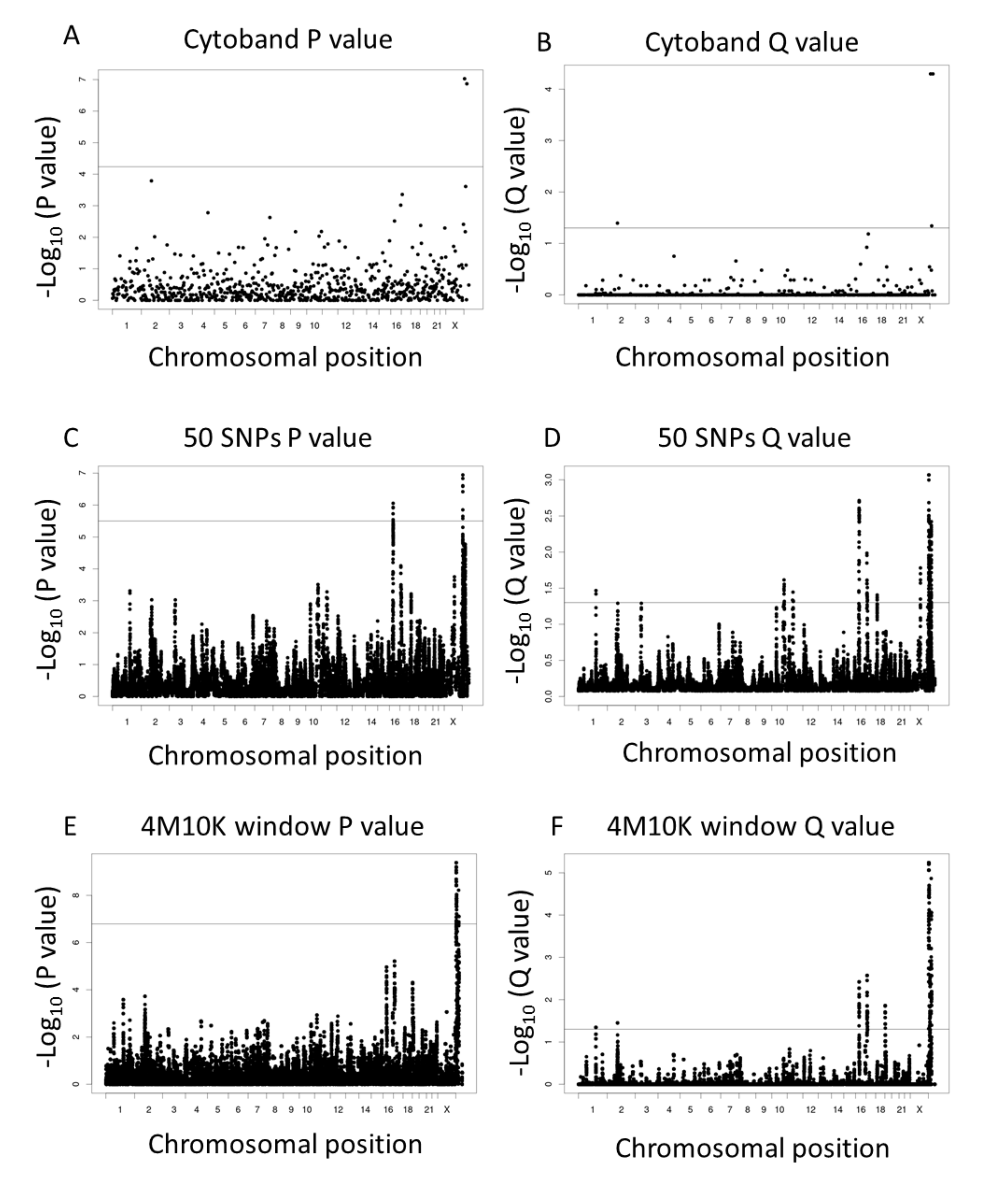
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsuo, T.; Yamane, T.; Ohtsuki, H. Heredity versus abnormalities in pregnancy and delivery as risk factors for different types of comitant strabismus. J. Pediatr. Ophthalmol. Strabismus 2001, 38, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Hayashi, M.; Fujiwara, H.; Yamane, T.; Ohtsuki, H. Concordance of strabismic phenotypes in monozygotic versus multizygotic twins and other multiple births. Jpn. J. Ophthalmol. 2002, 46, 59–64. [Google Scholar] [CrossRef]
- Maconachie, G.; Gottlob, I.; McLean, R.J. Risk factors and genetics in common comitant strabismus: A systematic review of the literature. JAMA Ophthalmol. 2013, 131, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Members of the Complex Trait Consortium. The nature and identification of quantitative trait loci: A community’s view. Nat. Rev. Genet. 2003, 4, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, L.; McKusick, V.A. Dissecting human disease in the postgenomic era. Science 2001, 291, 1224–1229. [Google Scholar] [CrossRef]
- Rice, A.; Nsengimana, J.; Simmons, I.G.; Toomes, C.; Hoole, J.; Willoughby, C.E.; Cassidy, F.; Williams, G.A.; George, N.D.; Sheridan, E.; et al. Replication of the recessive STBMS1 locus but with dominant inheritance. Investig. Opthalmol. Vis. Sci. 2009, 50, 3210–3217. [Google Scholar] [CrossRef]
- Parikh, V.; Shugart, Y.Y.; Doheny, K.F.; Zhang, J.; Li, L.; Williams, J.; Hayden, D.; Craig, B.; Capo, H.; Chamblee, D.; et al. A strabismus susceptibility locus on chromosome 7p. Proc. Natl. Acad. Sci. USA 2003, 100, 12283–12288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Matsuo, T. MGST2 and WNT2 are candidate genes for comitant strabismus susceptibility in Japanese patients. PeerJ 2017, 5, e3935. [Google Scholar] [CrossRef] [Green Version]
- Shaaban, S.; Matsuo, T.; Fujiwara, H.; Itoshima, E.; Furuse, T.; Hasebe, S.; Zhang, Q.; Ott, J.; Ohtsuki, H. Chromosomes 4q28.3 and 7q31.2 as new susceptibility loci for comitant strabismus. Investig. Opthalmol. Vis. Sci. 2009, 50, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Magwene, P.M.; Willis, J.H.; Kelly, J.K. The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput. Biol. 2011, 7, e1002255. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.; Mangano, E.; Barizzone, N.; Bordoni, R.; Sorosina, M.; Clarelli, F.; Corrado, L.; Boneschi, F.M.; D’Alfonso, S.; De Bellis, G. Next generation sequencing of pooled samples: Guideline for variants’ filtering. Sci. Rep. 2016, 6, 33735. [Google Scholar] [CrossRef]
- Terauchi, R.; Abe, A.; Takagi, H.; Tamiru, M.; Fekih, R.; Natsume, S.; Yaegashi, H.; Kosugi, S.; Kanzaki, H.; Matsumura, H.; et al. Whole genome sequencing to identify genes and QTL in rice. In Advances in the Understanding of Biological Sciences Using Next Generation Sequencing (NGS) Approaches; Springer: Cham, Switzerland, 2015; pp. 33–42. [Google Scholar] [CrossRef]
- Qanbari, S.; Strom, T.M.; Haberer, G.; Weigend, S.; Gheyas, A.A.; Turner, F.; Burt, D.W.; Preisinger, R.; Gianola, D.; Simianer, H. A high resolution genome-wide scan for significant selective sweeps: An application to pooled sequence data in laying chickens. PLoS ONE 2012, 7, e49525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, M.; Foucaud, J.; Gharbi, K.; Cézard, T.; Galan, M.; Loiseau, A.; Thomson, M.; Pudlo, P.; Kerdelhué, C.; Estoup, A. Estimation of population allele frequencies from next-generation sequencing data: Pool-versus individual-based genotyping. Mol. Ecol. 2013, 22, 3766–3779. [Google Scholar] [CrossRef]
- Zeeland, A.A.S.-V.; Bloss, C.; Tewhey, R.; Bansal, V.; Torkamani, A.; Libiger, O.; Duvvuri, V.; Wineinger, N.; Galvez, L.; Darst, B.F.; et al. Evidence for the role of EP HX2 gene variants in anorexia nervosa. Mol. Psychiatry 2014, 19, 724–732. [Google Scholar] [CrossRef]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef]
- Ramos, E.; Levinson, B.T.; Chasnoff, S.; Hughes, A.; Young, A.L.; Thornton, K.; Li, A.; Vallania, F.L.; Province, M.; Druley, T.E. Population-based rare variant detection via pooled exome or custom hybridization capture with or without individual indexing. BMC Genom. 2012, 13, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niranjan, T.S.; Adamczyk, A.; Bravo, H.C.; Taub, M.A.; Wheelan, S.J.; Irizarry, R.; Wang, T. Effective detection of rare variants in pooled DNA samples using cross-pool tail-curve analysis. Genome Biol. 2011, 12, P45. [Google Scholar] [CrossRef]
- Liu, Q.; Shen, E.; Min, Q.; Li, X.; Wang, X.; Li, X.; Sun, Z.S.; Wu, J. Exome-assistant: A rapid and easy detection of disease-related genes and genetic variations from exome sequencing. BMC Genom. 2012, 13, 692. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Koyutürk, M.; Maxwell, S.; Xiang, M.; Veigl, M.; Cooper, R.S.; Tayo, B.O.; Li, L.; LaFramboise, T.; Wang, Z.; et al. Discovery of common sequences absent in the human reference genome using pooled samples from next generation sequencing. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Hisano, H.; Sakamoto, K.; Takagi, H.; Terauchi, R.; Sato, K. Exome QTL-seq maps monogenic locus and QTLs in barley. BMC Genom. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosugi, S.; Natsume, S.; Yoshida, K.; MacLean, D.; Cano, L.; Kamoun, S.; Terauchi, R. Coval: Improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS ONE 2013, 8, e75402. [Google Scholar] [CrossRef] [Green Version]
- Downes, K.; Barratt, B.J.; Akan, P.; Bumpstead, S.J.; Taylor, S.D.; Clayton, D.G.; Deloukas, P. SNP allele frequency estimation in DNA pools and variance components analysis. BioTechniques 2004, 36, 840–845. [Google Scholar] [CrossRef]
- Vitti, J.J.; Grossman, S.R.; Sabeti, P.C. Detecting natural selection in genomic data. Annu. Rev. Genet. 2013, 47, 97–120. [Google Scholar] [CrossRef]
- Hong, E.P.; Park, J.W. Sample size and statistical power calculation in genetic association studies. Genom. Inform. 2012, 10, 117–122. [Google Scholar] [CrossRef]
- Huang, H.W.; Mullikin, J.C.; Hansen, N.F.; NISC Comparative Sequencing Program. Evaluation of variant detection software for pooled next-generation sequence data. BMC Bioinform. 2015, 16, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L. Haplotype phasing: Existing methods and new developments. Nat. Rev. Genet. 2011, 12, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xue, X.; Xie, X.; Ye, K.; Zhu, X.; Elston, R.C. Adjustment for covariates using summary statistics of genome-wide association studies. Genet. Epidemiol. 2018, 42, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.H.; Martin, L.S.; Eskin, E. Population structure in genetic studies: Confounding factors and mixed models. PLoS Genet. 2018, 14, e1007309. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.-J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef]
- Shaaban, S.; Matsuo, T.; Strauch, K.; Ohtsuki, H. Investigation of parent-of-origin effect in comitant strabismus using MOD score analysis. Mol. Vis. 2009, 15, 1351–1358. [Google Scholar] [PubMed]
- Khan, A.O.; Shinwari, J.; Abu Dhaim, N.; Khalil, D.; Al Sharif, L.; Al Tassan, N. Potential linkage of different phenotypic forms of childhood strabismus to a recessive susceptibility locus (16p13.12-p12.3). Mol. Vis. 2011, 17, 971–976. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Pairs | Esotropia (ET)/Normal (N) | Exotropia (XT)/Normal (N) | ET + XT/N | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ET | N | ET/N | XT | N | XT/N | ET+XT | N | ET + XT/N | |
| The number of SNP call co6cov3 (co = 6, depth ≥ 3) | 83,732 | 83,732 | 83,732 | 83,107 | 83,107 | 83,107 | 98,730 | 98,730 | 98,730 |
| SNPs with depth ≥ 20 | 16,383 | 15,897 | 12,717 | 15,905 | 15,678 | 12,473 | 33,204 | 13,851 | 13,711 |
| SNP index ≥ 0.3 | 15,946 | 15,514 | 12,242 | 15,576 | 15,365 | 12,108 | 32,108 | 13,707 | 13,367 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Matsuo, T.; Hamasaki, I.; Sato, K. Whole Exome-Sequencing of Pooled Genomic DNA Samples to Detect Quantitative Trait Loci in Esotropia and Exotropia of Strabismus in Japanese. Life 2022, 12, 41. https://doi.org/10.3390/life12010041
Zhang J, Matsuo T, Hamasaki I, Sato K. Whole Exome-Sequencing of Pooled Genomic DNA Samples to Detect Quantitative Trait Loci in Esotropia and Exotropia of Strabismus in Japanese. Life. 2022; 12(1):41. https://doi.org/10.3390/life12010041
Chicago/Turabian StyleZhang, Jingjing, Toshihiko Matsuo, Ichiro Hamasaki, and Kazuhiro Sato. 2022. "Whole Exome-Sequencing of Pooled Genomic DNA Samples to Detect Quantitative Trait Loci in Esotropia and Exotropia of Strabismus in Japanese" Life 12, no. 1: 41. https://doi.org/10.3390/life12010041
APA StyleZhang, J., Matsuo, T., Hamasaki, I., & Sato, K. (2022). Whole Exome-Sequencing of Pooled Genomic DNA Samples to Detect Quantitative Trait Loci in Esotropia and Exotropia of Strabismus in Japanese. Life, 12(1), 41. https://doi.org/10.3390/life12010041