
Narrow Precursor Mass Range for DIA–MS Enhances Protein Identification and Quantification in Arabidopsis


Abstract
1. Introduction
2. Materials and Methods
2.1. Arabidopsis Root Cell Suspension Culture
2.2. Protein Extraction and Digestion
2.3. Mass Spectrometric Analysis Using Data Independent Acquisition (DIA) Mode
2.4. DIA–MS Data Analysis Using Spectronaut
2.4.1. Library Generation
2.4.2. Quantitation Analysis
3. Results
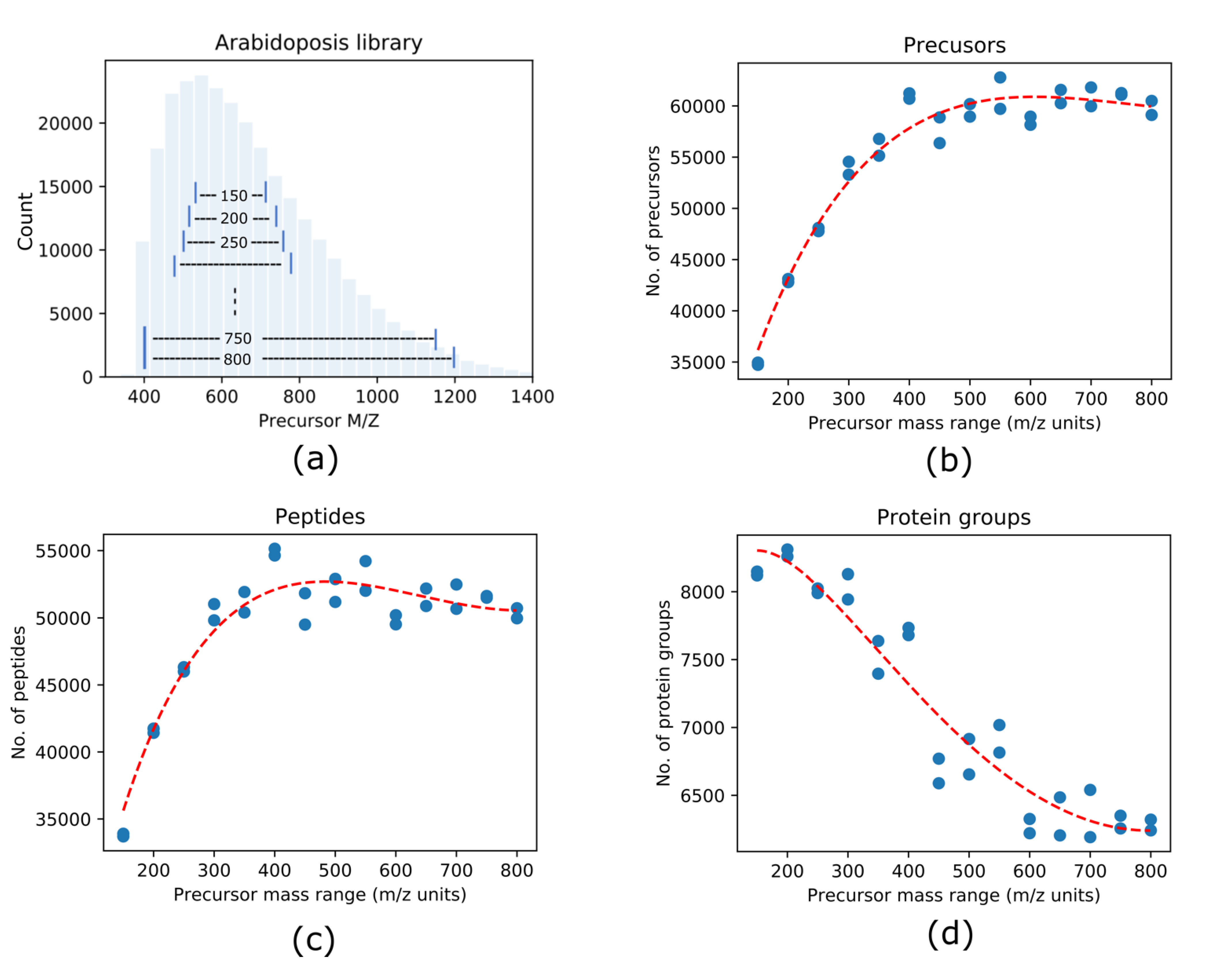
3.1. Effects of DIA Precursor Mass Range Selection on the Protein Identification
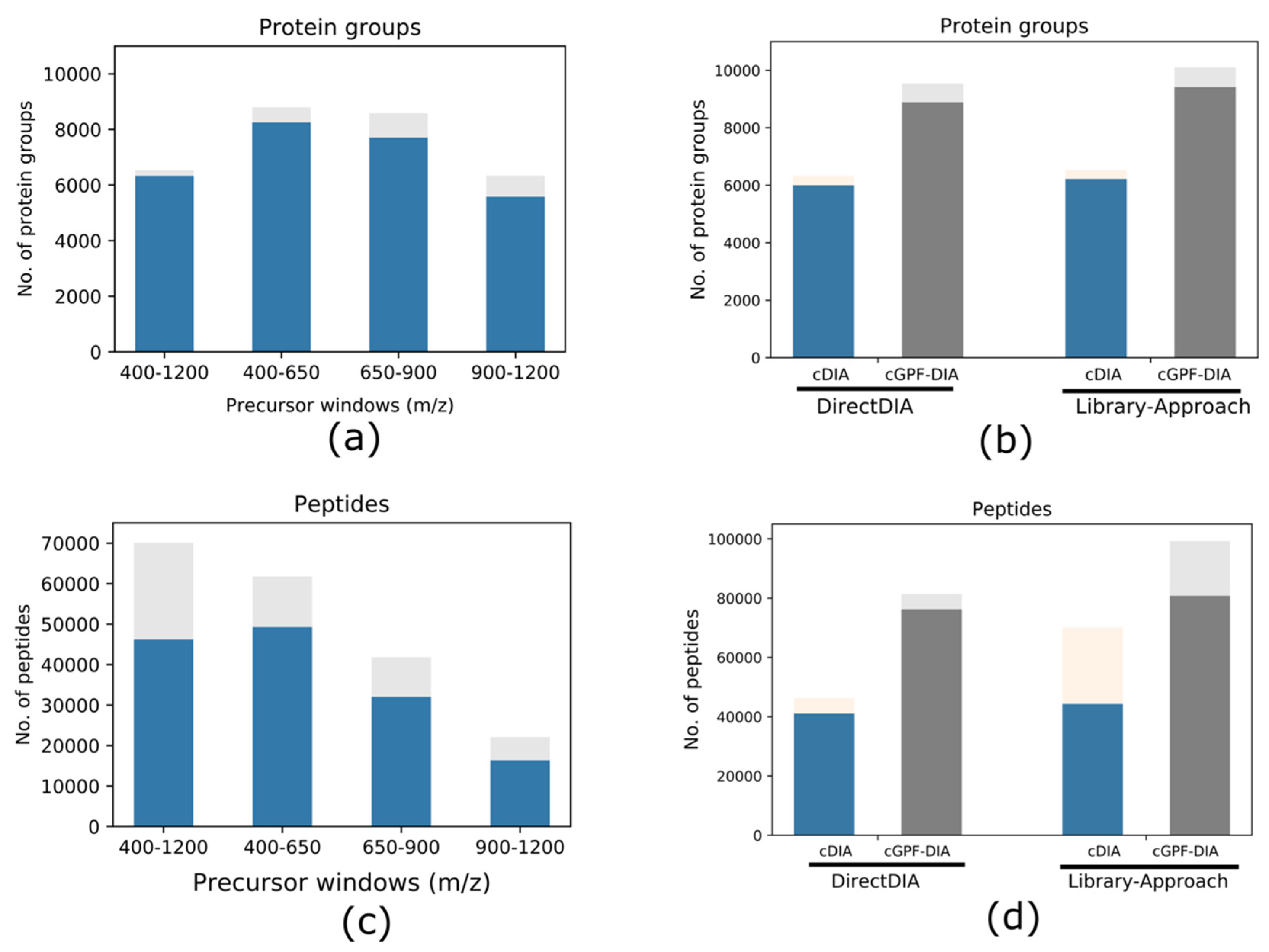
3.2. Narrow Precursor Scan Range Quantified Higher Number of Proteins
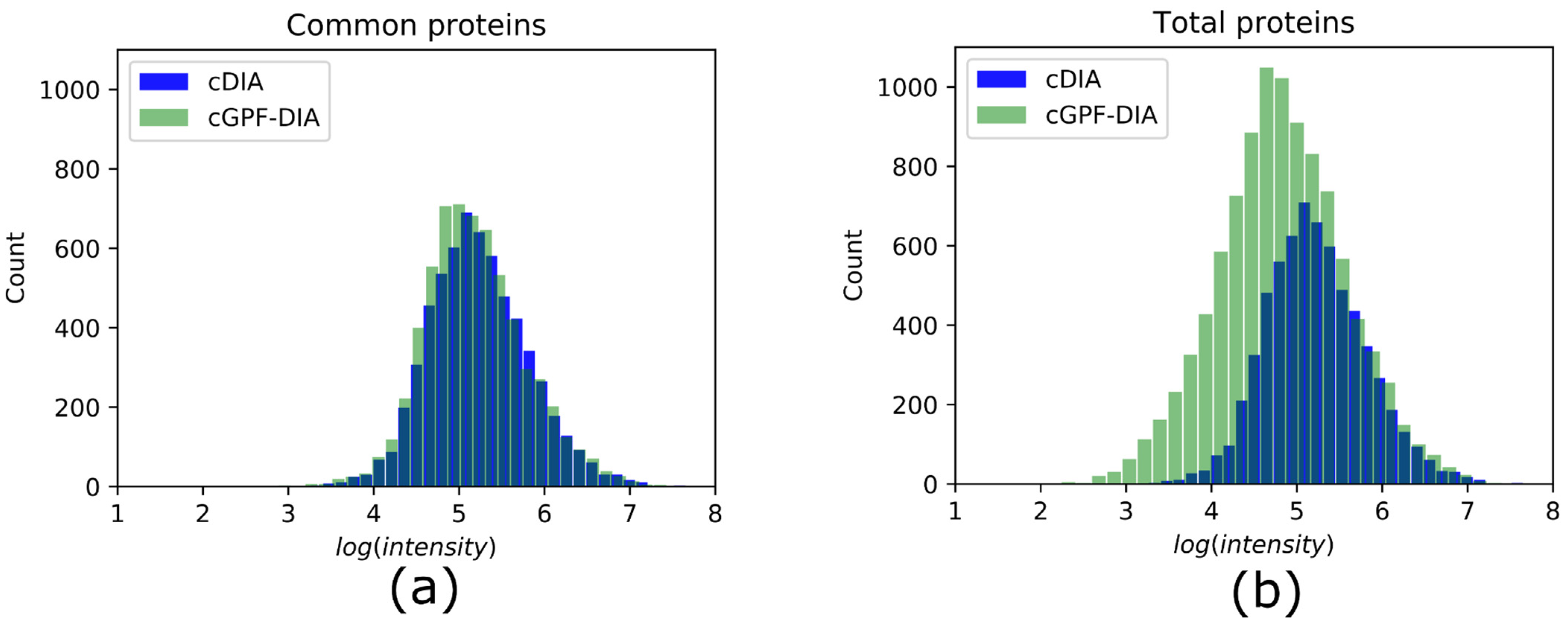
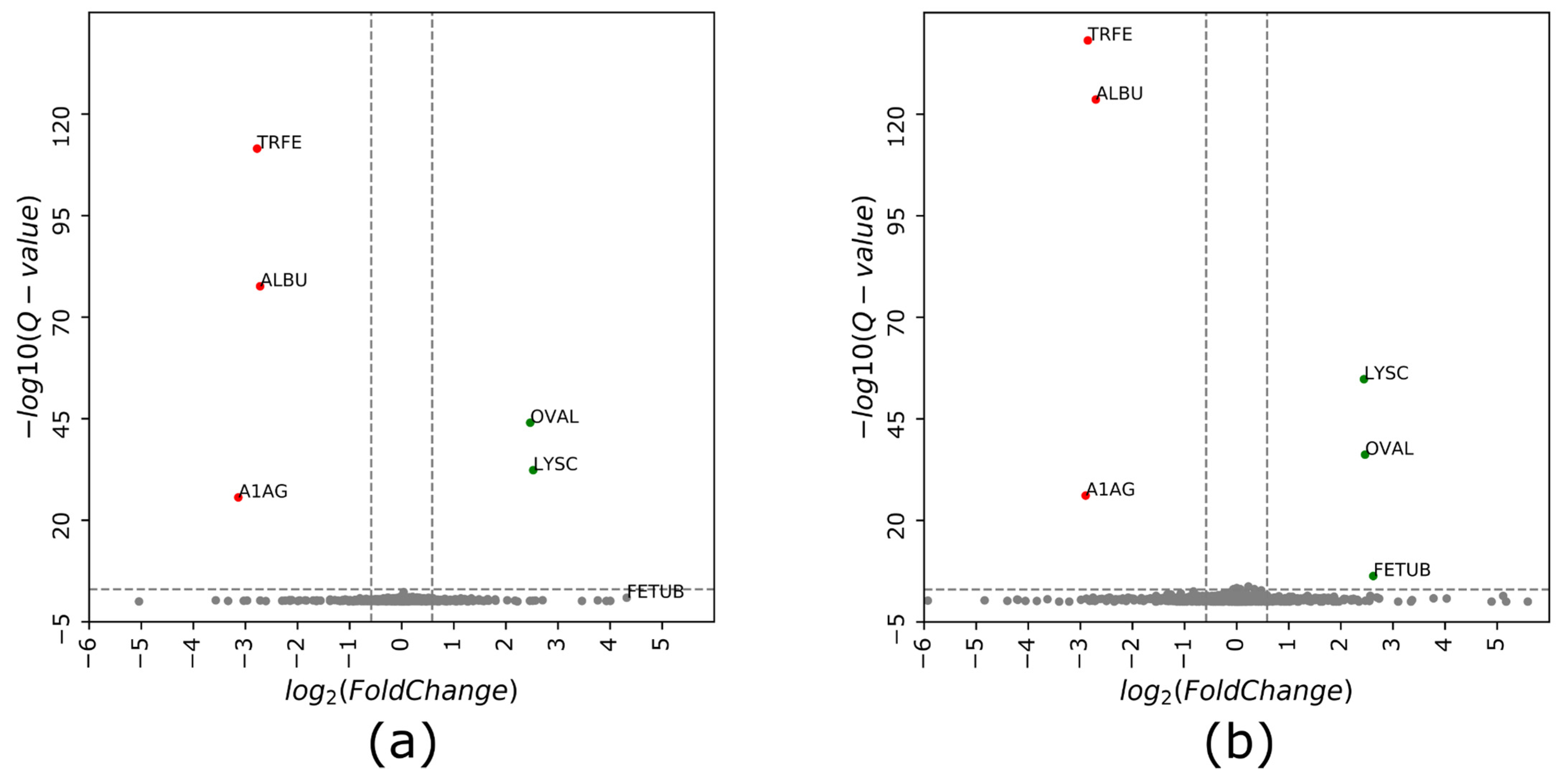
3.3. cGPF-DIA Recovered Low Abundance of Proteins
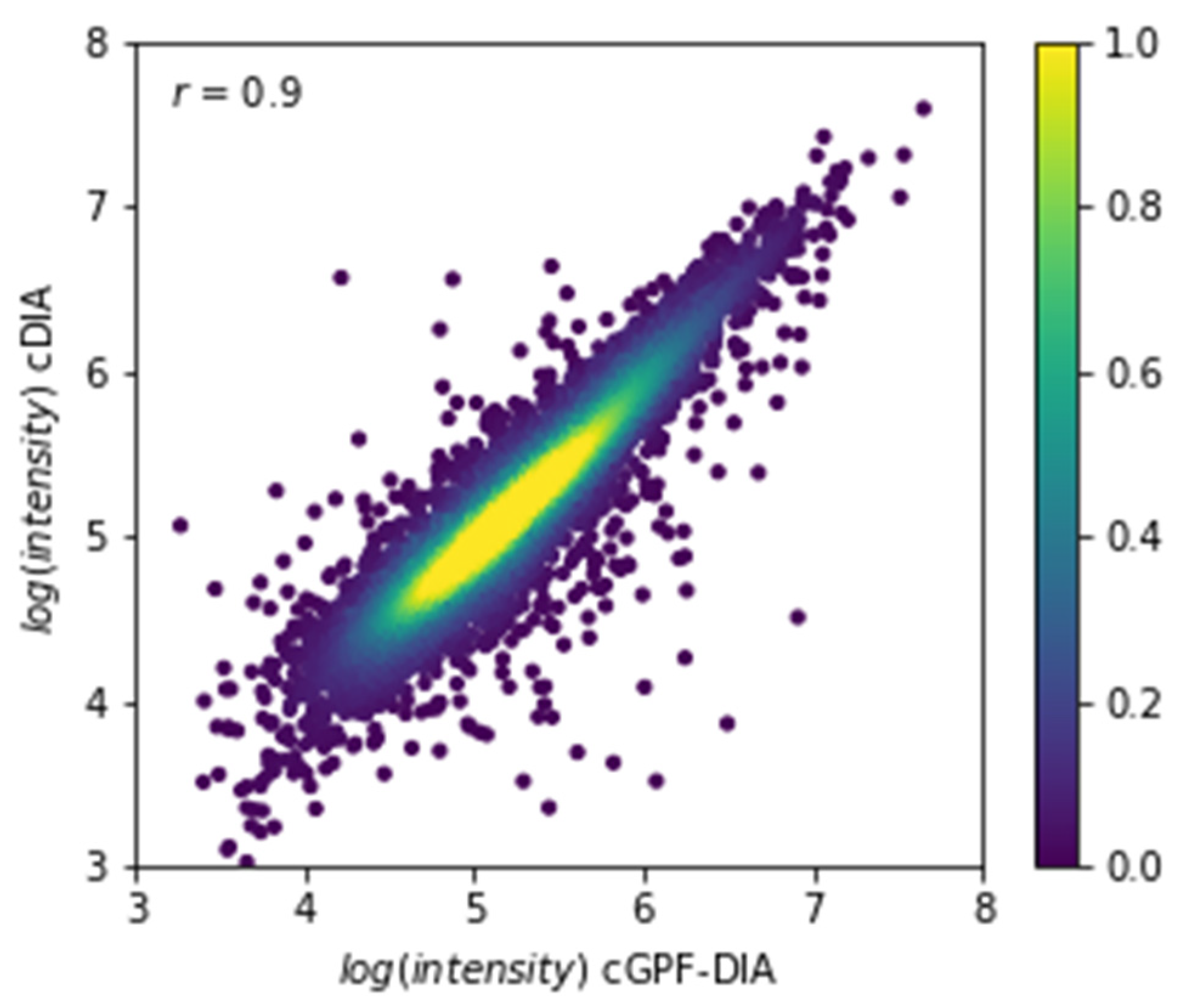
3.4. Narrow Precursor Ranges DIA Enabled Accurate Quantitation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, H.; Wong, W.C.; Sem, X.; Han, H.; Ong, S.M.; Tan, Y.C.; Yeap, W.H.; Gan, C.S.; Ng, K.Q.; et al. Identification of novel functional differences in monocyte subsets using proteomic and transcriptomic methods. J. Proteome Res. 2009, 8, 4028–4038. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Zhang, H.; Liu, P.; Tsao, S.W.; Lung, M.L.; Mak, N.K.; Ngok-Shun Wong, R.; Ying-Kit Yue, P. Proteomic analysis of exosomes from nasopharyngeal carcinoma cell identifies intercellular transfer of angiogenic proteins. Int. J. Cancer 2015, 137, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Jin Kim, Y.; Heidbrink, J.L.; Moore, P.A.; Ruben, S.M. Drug target identification and quantitative proteomics. Expert Opin. Drug Discov. 2006, 1, 477–489. [Google Scholar] [CrossRef]
- Wang, J.; Wong, Y.K.; Zhang, J.; Lee, Y.M.; Hua, Z.C.; Shen, H.M.; Lin, Q. Drug Target Identification Using an iTRAQ-Based Quantitative Chemical Proteomics Approach-Based on a Target Profiling Study of Andrographolide. Methods Enzymol. 2017, 586, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Dashatan, N.; Ahmadi, N.; Rezaei-Tavirani, M.; Koushki, M. Identification of differential protein expression and putative drug target in metacyclic stage of Leishmania major and Leishmania tropica: A quantitative proteomics and computational view. Comp. Immunol. Microbiol. Infect. Dis. 2021, 75, 101617. [Google Scholar] [CrossRef]
- Unwin, R.D.; Pierce, A.; Watson, R.B.; Sternberg, D.W.; Whetton, A.D. Quantitative proteomic analysis using isobaric protein tags enables rapid comparison of changes in transcript and protein levels in transformed cells. Mol. Cell. Proteom. 2005, 4, 924–935. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Chelius, D.; Bondarenko, P.V. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res. 2002, 1, 317–323. [Google Scholar] [CrossRef]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. iTRAQ underestimation in simple and complex mixtures: The good, the bad and the ugly. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef]
- Kuhn, E.; Wu, J.; Karl, J.; Liao, H.; Zolg, W.; Guild, B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 2004, 4, 1175–1186. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Rost, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, P.; Guo, T.; Zhao, H.; Bensaddek, D.; Aebersold, R.; Xiong, L. Arabidopsis proteome and the mass spectral assay library. Sci. Data 2019, 6, 278. [Google Scholar] [CrossRef] [PubMed]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Xuan, Y.; Sondermann, J.; Schmidt, M.; Gomez-Varela, D.; Reiter, L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteom. 2017, 16, 2296–2309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC-SWATH-MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Amodei, D.; Egertson, J.; MacLean, B.X.; Johnson, R.; Merrihew, G.E.; Keller, A.; Marsh, D.; Vitek, O.; Mallick, P.; MacCoss, M.J. Improving Precursor Selectivity in Data-Independent Acquisition Using Overlapping Windows. J. Am. Soc. Mass Spectrom. 2019, 30, 669–684. [Google Scholar] [CrossRef]
- Borras, E.; Sabido, E. DIA+: A Data-Independent Acquisition Method Combining Multiple Precursor Charges to Improve Peptide Signal. Anal. Chem. 2018, 90, 12339–12341. [Google Scholar] [CrossRef]
- Cai, X.; Ge, W.; Yi, X.; Sun, R.; Zhu, J.; Lu, C.; Sun, P.; Zhu, T.; Ruan, G.; Yuan, C.; et al. PulseDIA: Data-Independent Acquisition Mass Spectrometry Using Multi-Injection Pulsed Gas-Phase Fractionation. J. Proteome Res. 2021, 20, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Liang, X.; Zhang, F.; Xu, L.; Xiang, N.; Sun, R.; Liu, W.; Xue, Z.; Yi, X.; Wang, B.; et al. Optimization of Spectral Library Size Improves DIA-MS Proteome Coverage. bioRxiv 2020. [CrossRef]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Kuster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1548. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Prasad, S.; Belford, M.W.; Bailey, D.J.; McAlister, G.C.; Abbatiello, S.E.; Huguet, R.; Wouters, E.R.; Dunyach, J.J.; Brademan, D.R.; et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 9529–9537. [Google Scholar] [CrossRef]
- Ordonez, N.M.; Marondedze, C.; Thomas, L.; Pasqualini, S.; Shabala, L.; Shabala, S.; Gehring, C. Cyclic mononucleotides modulate potassium and calcium flux responses to H2O2 in Arabidopsis roots. FEBS Lett. 2014, 588, 1008–1015. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qian, P.Y.; Ravasi, T. Selective phosphorylation during early macrophage differentiation. Proteomics 2015, 15, 3731–3743. [Google Scholar] [CrossRef][Green Version]
- Guan, S.; Taylor, P.P.; Han, Z.; Moran, M.F.; Ma, B. Data Dependent-Independent Acquisition (DDIA) Proteomics. J. Proteome Res. 2020, 19, 3230–3237. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.H.; Liu, L.C.; Tan, H.T.; Tan, K.; Ng, D.; Zhang, W.; Yang, Y.; Tate, S.; Bi, X. A comprehensive CHO SWATH-MS spectral library for robust quantitative profiling of 10,000 proteins. Sci. Data 2020, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Lou, R.; Tang, P.; Ding, K.; Li, S.; Tian, C.; Li, Y.; Zhao, S.; Zhang, Y.; Shui, W. Hybrid Spectral Library Combining DIA-MS Data and a Targeted Virtual Library Substantially Deepens the Proteome Coverage. iScience 2020, 23, 100903. [Google Scholar] [CrossRef] [PubMed]
- Pak, H.; Michaux, J.; Huber, F.; Chong, C.; Stevenson, B.J.; Muller, M.; Coukos, G.; Bassani-Sternberg, M. Sensitive Immunopeptidomics by Leveraging Available Large-Scale Multi-HLA Spectral Libraries, Data-Independent Acquisition, and MS/MS Prediction. Mol. Cell. Proteom. 2021, 20, 100080. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Information | Quantity (fmol) and Ratio of Spike-In | Quantitation by cDIA* | Quantitation by cGPF-DIA** | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SwissProt Accession | Molecular Weight (Da) | S1 | S2 | S1:S2 | S1:S2 | Q-value | S1:S2 | Q-value | |
| BSA | P02769 | 69300 | 64.1 | 8.0 | 8:1 | 6.6: 1 | 2.5 × 10−18 | 6.5: 1 | 2.4 × 10−12 |
| Transferrin | P02787 | 77070 | 57.7 | 7.2 | 8:1 | 6.8: 1 | 3.3 × 10−112 | 7.2: 1 | 7.0 × 10−13 |
| alpha-acid glycoprotein | Q3SZR3 | 23190 | 191.7 | 24.0 | 8:1 | 8.8: 1 | 2.4 × 10−26 | 7.5: 1 | 7.7 × 10−27 |
| Lysozyme | P00689 | 16240 | 34.2 | 171.0 | 1:5 | 1: 5.8 | 4.7 × 10−33 | 1: 5.4 | 1.7 × 10−55 |
| Fetuin | Q58D62 | 42670 | 13.0 | 65.1 | 1:5 | 1: 19.7 | 1.2 × 10−1 | 1: 6.1 | 4.9 × 10−7 |
| Ovalbumin | P01012 | 42890 | 13.0 | 64.8 | 1:5 | 1: 5.5 | 9.7 × 10−78 | 1: 5.5 | 6.6 × 10−37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Bensaddek, D. Narrow Precursor Mass Range for DIA–MS Enhances Protein Identification and Quantification in Arabidopsis. Life 2021, 11, 982. https://doi.org/10.3390/life11090982
Zhang H, Bensaddek D. Narrow Precursor Mass Range for DIA–MS Enhances Protein Identification and Quantification in Arabidopsis. Life. 2021; 11(9):982. https://doi.org/10.3390/life11090982
Chicago/Turabian StyleZhang, Huoming, and Dalila Bensaddek. 2021. "Narrow Precursor Mass Range for DIA–MS Enhances Protein Identification and Quantification in Arabidopsis" Life 11, no. 9: 982. https://doi.org/10.3390/life11090982
APA StyleZhang, H., & Bensaddek, D. (2021). Narrow Precursor Mass Range for DIA–MS Enhances Protein Identification and Quantification in Arabidopsis. Life, 11(9), 982. https://doi.org/10.3390/life11090982