
The Link between Type 2 Diabetes Mellitus and the Polymorphisms of Glutathione-Metabolizing Genes Suggests a New Hypothesis Explaining Disease Initiation and Progression



Abstract
1. Introduction
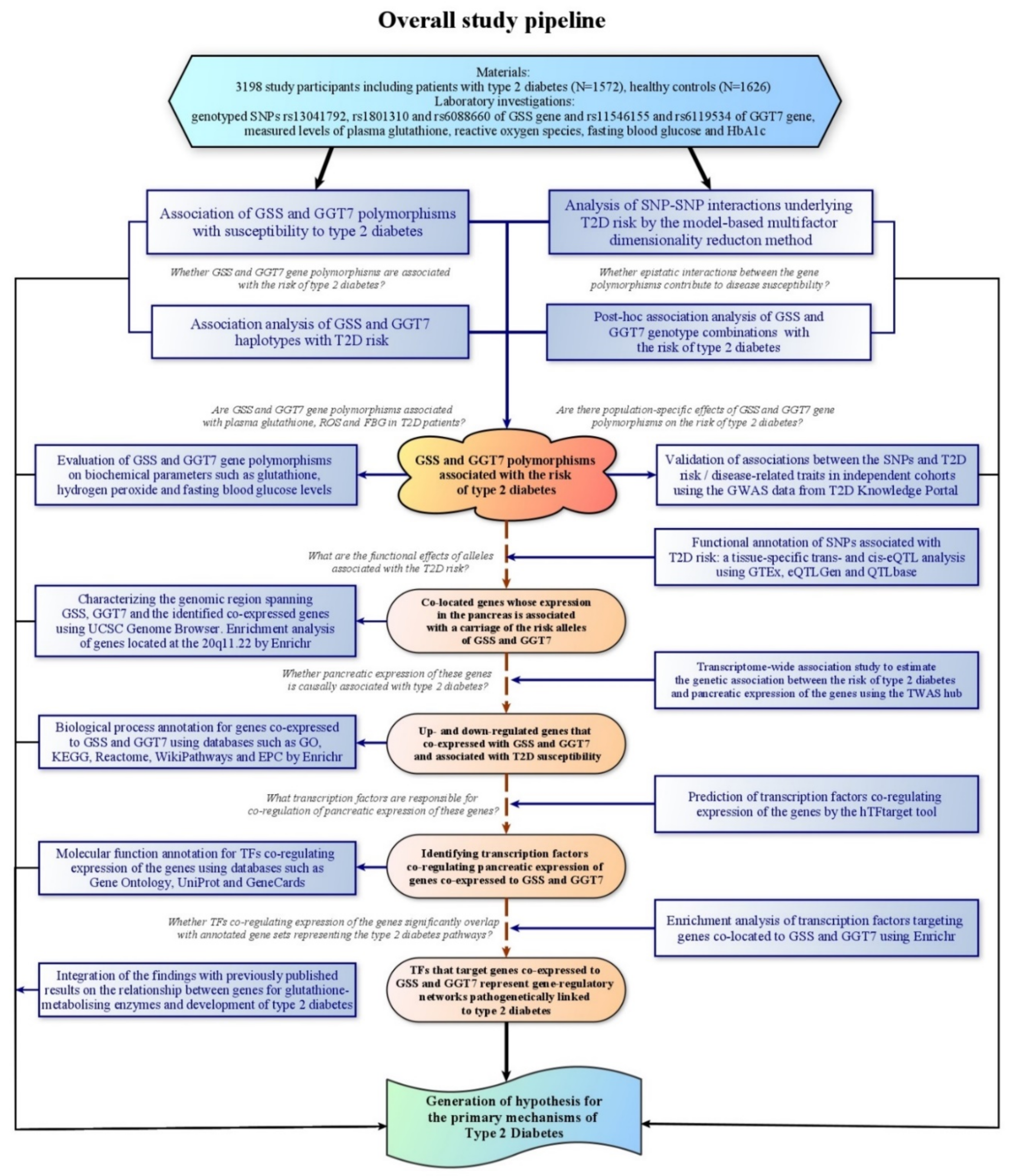
2. Materials and Methods
2.1. Study Participants and Diagnosis of Type 2 Diabetes
2.2. Blood Specimen Collection and Processing
2.3. SNP Selection and Molecular Genetic Analysis
2.4. Biochemical Analysis
2.5. Statistical Power Calculation
2.6. Association of GSS and GGT7 Gene Polymorphisms with Type 2 Diabetes
2.7. A Replication of the Associations in Independent Populations
2.8. Analysis of the Contribution of SNP–SNP Interactions to T2D Susceptibility
2.9. Impact of GSS and GGT7 Gene Polymorphisms on Biochemical Parameters
2.10. Functional Annotation of GSS and GGT7 Gene Polymorphisms
2.11. Transcriptome-Wide Association Analysis of Genes Co-Expressed with GSS and GGT7
2.12. In Silico Prediction of Transcription Factors Co-Regulating Expression of the Target Genes
2.13. Gene Set Enrichment Analysis
3. Results
3.1. Demographic, Clinical and Laboratory Characteristics of the Study Participants
3.2. Association of GSS and GGT7 Gene Polymorphisms with the Risk of T2D
3.3. Associations of GSS and GGT7 Haplotypes with Disease Susceptibility
3.4. Epistatic Interactions between the Polymorphisms and Susceptibility to Type 2 Diabetes
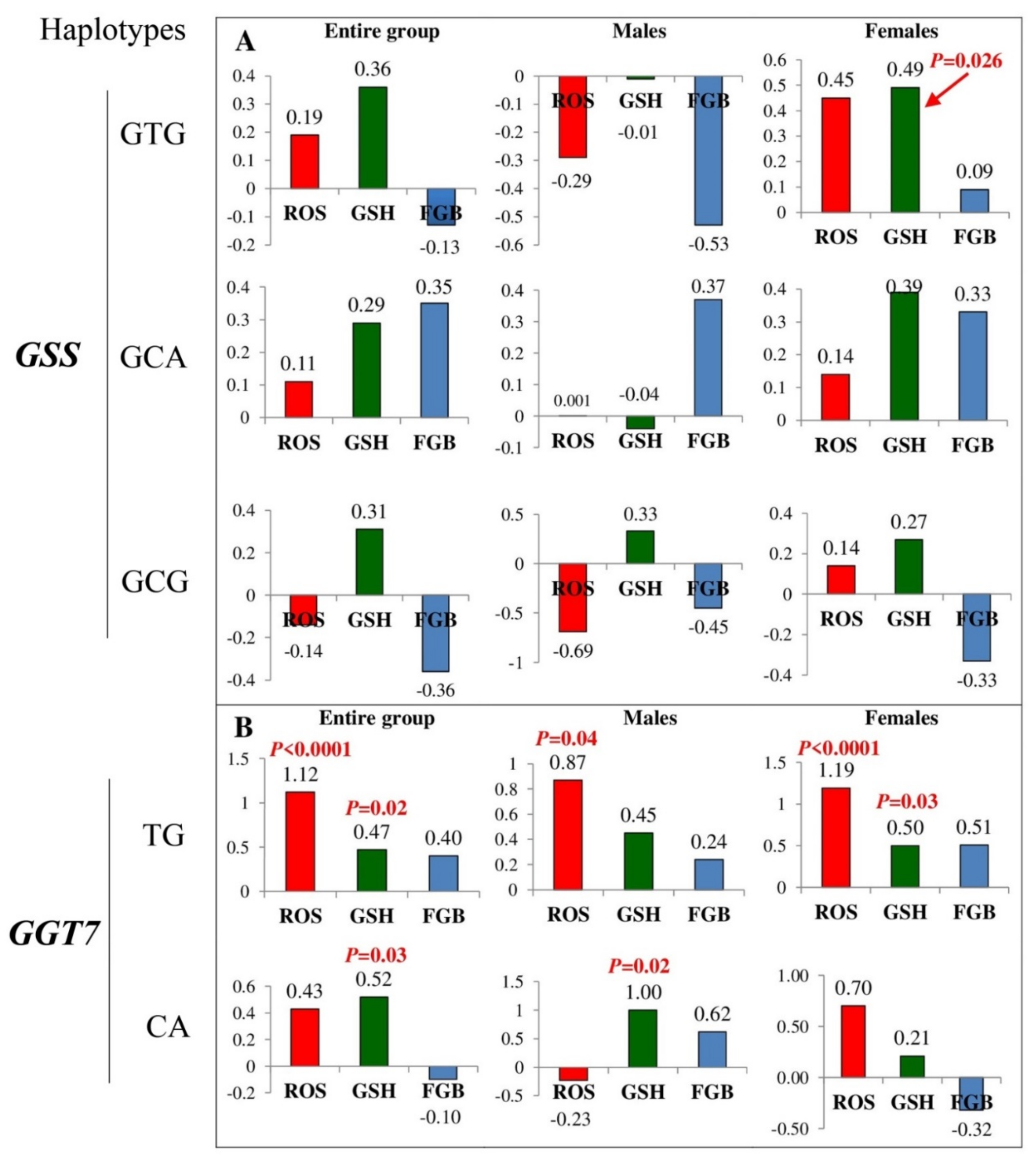
3.5. Influence of GSS and GGT7 Gene Polymorphisms on Biochemical Parameters
3.6. Validation for the Observed SNP-Disease Associations in Independent Populations
3.7. Functional Annotation of GSS and GGT7 Gene Polymorphisms
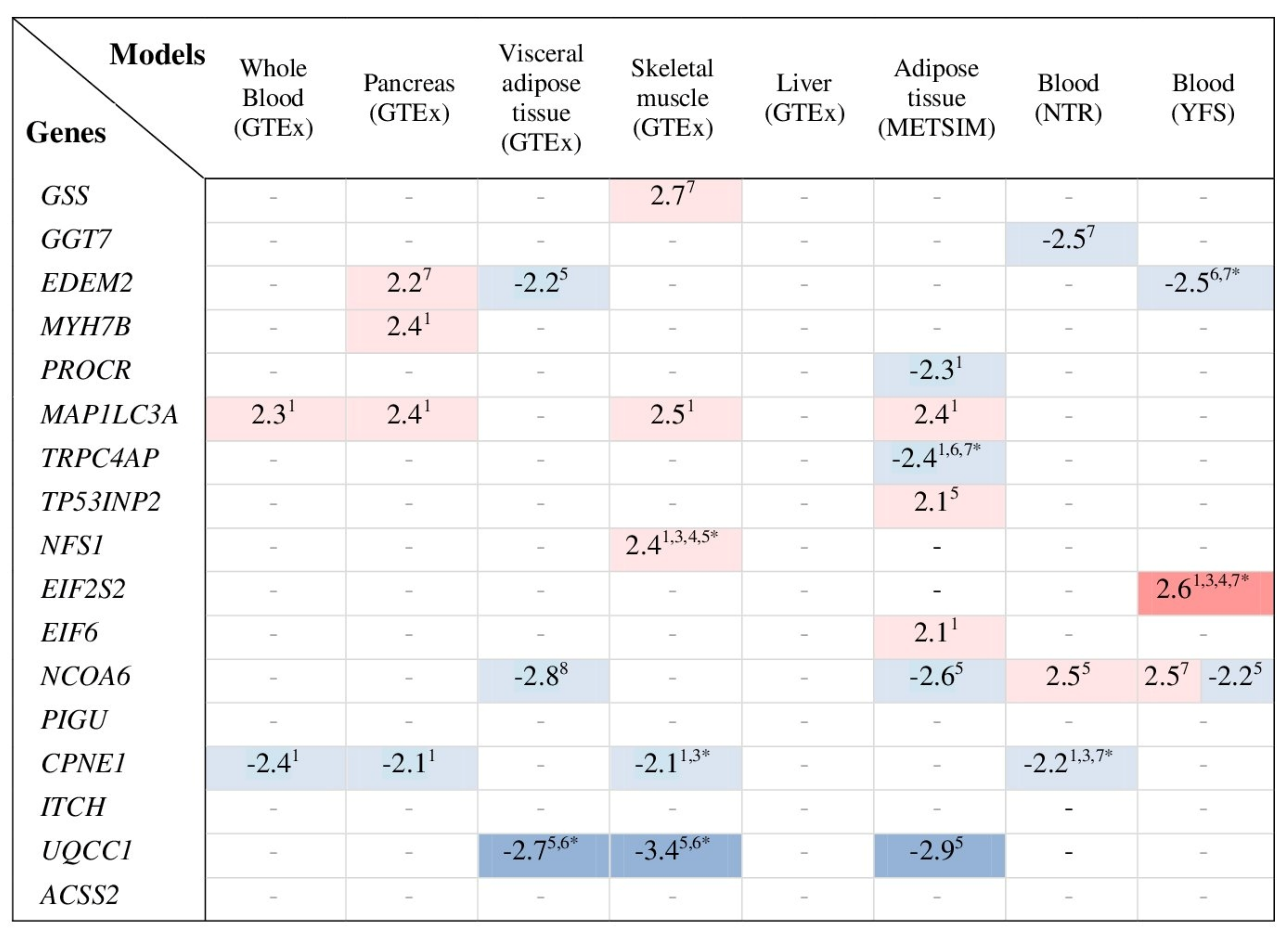
3.8. Transcriptome-Wide Association Study for Genes Co-Expressed with GSS and GGT7
3.9. Prediction of Transcription Factors Co-Regulating Expression of T2D Associated Genes and Their Enrichment Analysis
4. Discussion
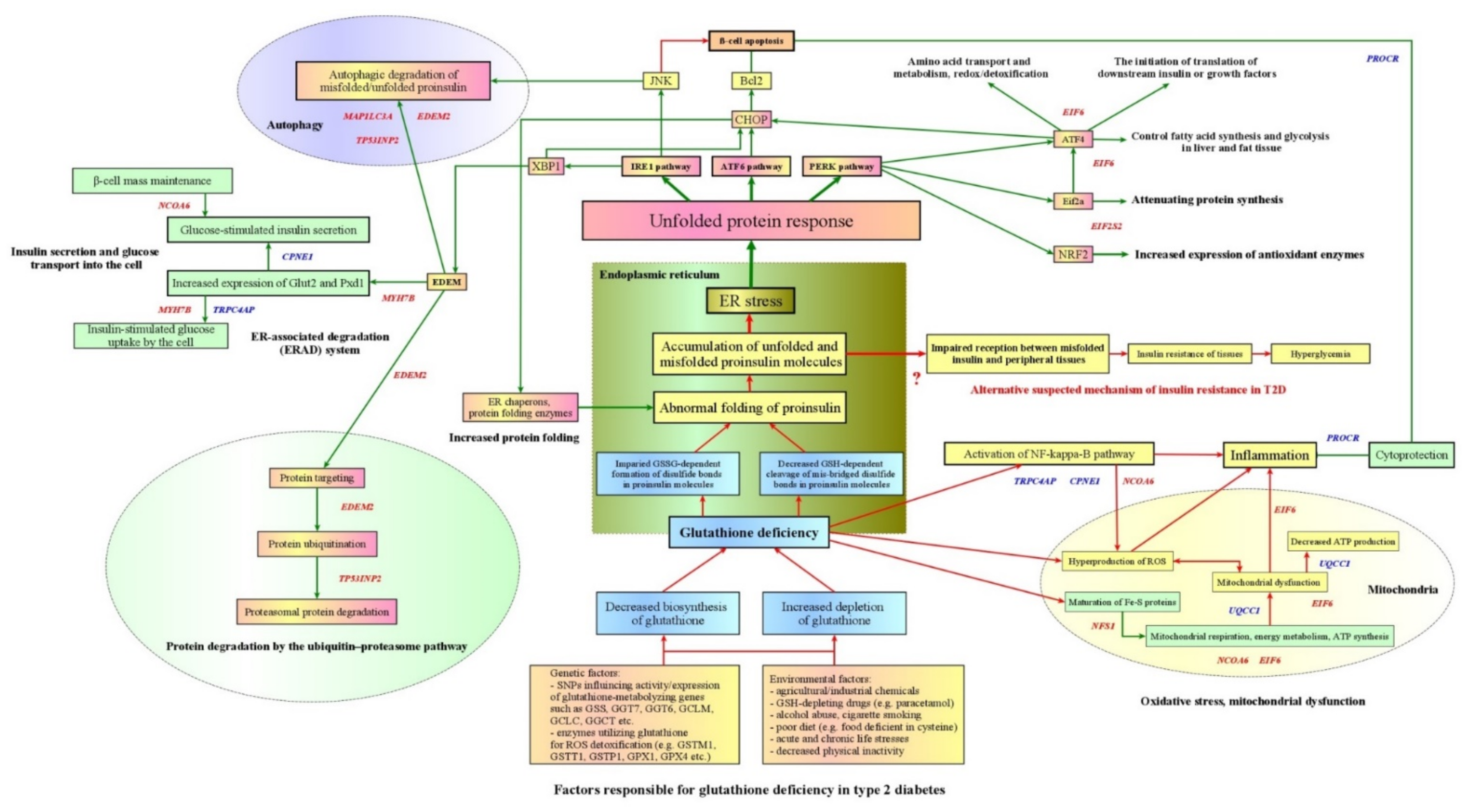
4.1. Overview of the Main Findings
4.2. Genetic and Environmental Factors Responsible for Endogenous Deficiency of Glutathione in Type 2 Diabetes
4.3. Glutathione Deficiency as a Cause of Impaired Folding of Proinsulin in Type 2 Diabetes
4.4. Study Limitations
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Dedov, I.; Shestakova, M.V.; Benedetti, M.M.; Simon, D.; Pakhomov, I.; Galstyan, G. Prevalence of type 2 diabetes mellitus (T2DM) in the adult Russian population (NATION study). Diabetes Res. Clin. Pract. 2016, 115, 90–95. [Google Scholar] [CrossRef]
- Sinclair, A.; Saeedi, P.; Kaundal, A.; Karuranga, S.; Malanda, B.; Williams, R. Diabetes and global ageing among 65–99-year-old adults: Findings from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108078. [Google Scholar] [CrossRef]
- Van Tilburg, J.; Van Haeften, T.W.; Pearson, P.; Wijmenga, C. Defining the genetic contribution of type 2 diabetes mellitus. J. Med. Genet. 2001, 38, 569–578. [Google Scholar] [CrossRef]
- McCarthy, M.; Menzel, S. The genetics of type 2 diabetes. Br. J. Clin. Pharmacol. 2001, 51, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Frau, F.; Crowther, D.; Ruetten, H.; Allebrandt, K.V. Type-2 diabetes-associated variants with cross-trait relevance: Post-GWAs strategies for biological function interpretation. Mol. Genet. Metab. 2017, 121, 43–50. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017, 60, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, E.; Triplitt, C.; Solis-Herrera, C.; Mandarino, L.J.; DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Hayden, M.R.; Sowers, J.R. Redox Imbalance in Diabetes. Antioxid. Redox Signal. 2007, 9, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef]
- Murotomi, K.; Umeno, A.; Yasunaga, M.; Shichiri, M.; Ishida, N.; Abe, H.; Yoshida, Y.; Nakajima, Y. Switching from singlet-oxygen-mediated oxidation to free-radical-mediated oxidation in the pathogenesis of type 2 diabetes in model mouse. Free Radic. Res. 2014, 49, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H. Mechanism of Generation of Oxidative Stress and Pathophysiology of Type 2 Diabetes Mellitus: How Are They Interlinked? J. Cell. Biochem. 2017, 118, 3577–3585. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Lodovici, M. Circulating Oxidative Stress Biomarkers in Clinical Studies on Type 2 Diabetes and Its Complications. Oxidative Med. Cell. Longev. 2019, 2019, 5953685. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 2018, 45, 152–168. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Glutathione deficiency induces epigenetic alterations of vitamin D metabolism genes in the livers of high-fat diet-fed obese mice. Sci. Rep. 2019, 9, 14784. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Pallardó, F.V. Maintenance of glutathione levels and its importance in epigenetic regulation. Front. Pharmacol. 2014, 5, 88. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a Marker for Human Disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar] [CrossRef]
- Seltzer, H.S. Blood glutathione in mild diabetes mellitus before treatment and during sulfonylurea-induced hypoglycemia. Proc. Soc. Exp. Biol. Med. 1957, 95, 74–76. [Google Scholar] [CrossRef]
- McLennan, S.V.; Heffernan, S.; Wright, L.; Rae, C.; Fisher, E.; Yue, D.K.; Turtle, J.R. Changes in Hepatic Glutathione Metabolism in Diabetes. Diabetes 1991, 40, 344–348. [Google Scholar] [CrossRef]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Nitti, M.; Marengo, B.; Domenicotti, C.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A. Impaired synthesis contributes to diabetes-induced decrease in liver glutathione. Int. J. Mol. Med. 2012, 29, 899–905. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kalkan, I.H.; Suher, M. The relationship between the level of glutathione, impairment of glucose metabolism and complications of diabetes mellitus. Pak. J. Med. Sci. 2013, 29, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef]
- Azarova, I.; Bushueva, O.; Konoplya, A.; Polonikov, A. Glutathione S-transferase genes and the risk of type 2 diabetes mellitus: Role of sexual dimorphism, gene-gene and gene-smoking interactions in disease susceptibility. J. Diabetes 2018, 10, 398–407. [Google Scholar] [CrossRef]
- Azarova, I.; Klyosova, E.; Lazarenko, V.; Konoplya, A.; Polonikov, A. Genetic variants in glutamate cysteine ligase confer protection against type 2 diabetes. Mol. Biol. Rep. 2020, 47, 5793–5805. [Google Scholar] [CrossRef]
- Azarova, I.E.; Klyosova, E.Y.; Sakali, S.Y.; Kovalev, A.P. Contribution of rs11927381 polymorphism of the IGF2BP2 gene to the pathogenesis of type 2 diabetes. Res. Results Biomed. 2020, 6, 9–19. [Google Scholar] [CrossRef]
- Azarova, I.E.; Klyosova, E.Y.; Churilin, M.I.; Samgina, T.A.; Konoplya, A.I.; Polonikov, A.V. Genetic and biochemical investigation of the gamma-glutamylcyclotransferase role in predisposition to type 2 diabetes mellitus. Ecol. Genet. 2020, 18, 215–228. [Google Scholar] [CrossRef]
- Azarova, I.; Klyosova, E.; Shkurat, E.; Polonikov, A. Polymorphic Variants in gamma-glutamyltransferase 6 as New Genetic Markers of Type 2 Diabetes Mellitus. Metabolism 2021, 116, 154537. [Google Scholar] [CrossRef]
- Pinheiro, D.; Rocha-Filho, C.R.; Mundim, C.A.; Júnior, P.D.M.; Ulhoa, C.; Reis, A.A.S.; Ghedini, P. Evaluation of Glutathione S-Transferase GSTM1 and GSTT1 Deletion Polymorphisms on Type-2 Diabetes Mellitus Risk. PLoS ONE 2013, 8, e76262. [Google Scholar] [CrossRef]
- Chen, H.; Yu, M.; Li, M.; Zhao, R.; Zhu, Q.; Zhou, W.; Lu, M.; Lu, Y.; Zheng, T.; Jiang, J.; et al. Polymorphic variations in manganese superoxide dismutase (MnSOD), glutathione peroxidase-1 (GPX1), and catalase (CAT) contribute to elevated plasma triglyceride levels in Chinese patients with type 2 diabetes or diabetic cardiovascular disease. Mol. Cell. Biochem. 2011, 363, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Vats, P.; Sagar, N.; Singh, T.P.; Banerjee, M. Association ofSuperoxide dismutases(SOD1 and SOD2) andGlutathione peroxidase 1 (GPx1)gene polymorphisms with Type 2 diabetes mellitus. Free Radic. Res. 2014, 49, 17–24. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Polonikov, A.V.; Ivanov, V.P.; Belugin, D.A.; Khoroshaya, I.V.; Kolchanova, I.O.; Solodilova, M.A.; Tutochkina, M.P.; Stepchenko, A.A. Analysis of common transforming growth factor beta-1 gene polymorphisms in gastric and duodenal ulcer disease: Pilot study. J. Gastroenterol. Hepatol. 2007, 22, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A.; Vialykh, E.; Vasil’Eva, O.; Bulgakova, I.; Bushueva, O.; Illig, T.; Solodilova, M. Genetic Variation in Glutathione S-Transferase Genes and Risk of Nonfatal Cerebral Stroke in Patients Suffering from Essential Hypertension. J. Mol. Neurosci. 2012, 47, 511–513. [Google Scholar] [CrossRef]
- Polonikov, A.; Bushueva, O.Y.; Bulgakova, I.V.; Freidin, M.B.; Churnosov, M.; Solodilova, M.A.; Shvetsov, Y.D.; Ivanov, V.P. A comprehensive contribution of genes for aryl hydrocarbon receptor signaling pathway to hypertension susceptibility. Pharmacogenet. Genom. 2017, 27, 57–69. [Google Scholar] [CrossRef][Green Version]
- Xu, Z.; Taylor, J.A. SNPinfo: Integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009, 37, W600–W605. [Google Scholar] [CrossRef]
- Breton, C.V.; Salam, M.; Vora, H.; Gauderman, W.J.; Gilliland, F.D. Genetic Variation in the Glutathione Synthesis Pathway, Air Pollution, and Children’s Lung Function Growth. Am. J. Respir. Crit. Care Med. 2011, 183, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Abecasis, G.R. GAS Power Calculator: Web-based power calculator for genetic association studies. bioRxiv 2017, 164343. [Google Scholar] [CrossRef]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef] [PubMed]
- Kraft, P.; Zeggini, E.; Ioannidis, J.P.A. Replication in Genome-Wide Association Studies. Stat. Sci. 2009, 24, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.S.; Penrod, N.M.; Williams, S.M.; Moore, J.H. Failure to Replicate a Genetic Association May Provide Important Clues About Genetic Architecture. PLoS ONE 2009, 4, e5639. [Google Scholar] [CrossRef]
- Calle, M.L.; Urrea, V.; Vellalta, G.; Malats, N.; Steen, K.V. Improving strategies for detecting genetic patterns of disease susceptibility in association studies. Stat. Med. 2008, 27, 6532–6546. [Google Scholar] [CrossRef]
- Calle, M.L.; Urrea, V.; Malats, N.; Van Steen, K. mbmdr: An R package for exploring gene–gene interactions associated with binary or quantitative traits. Bioinformatics 2010, 26, 2198–2199. [Google Scholar] [CrossRef][Green Version]
- The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Kasela, S.; et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018, 447367. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, D.; Wang, J.; Zhao, K.; Zhou, Y.; Guo, Z.; Zhai, S.; Xu, H.; Cui, H.; Yao, H.; et al. QTLbase: An integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2019, 48, D983–D991. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Dai, J.; Jin, S.; Zhou, X. Aggregating multiple expression prediction models improves the power of transcriptome-wide association studies. Hum. Mol. Genet. 2021, 30, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; De Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Wu, Y.; Pan, C.; Raulerson, C.K.; Ko, A.; He, A.; Tilford, C.; Saleem, N.K.; Stančáková, A.; Scott, L.J.; et al. Genetic Regulation of Adipose Gene Expression and Cardio-Metabolic Traits. Am. J. Hum. Genet. 2017, 100, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; de Geus, E.J.; Vink, J.M.; Stubbe, J.H.; Distel, M.A.; Hottenga, J.J.; Posthuma, D.; van Beijsterveldt, T.C.; Hudziak, J.J.; Bartels, M.; et al. Netherlands Twin Register: From twins to twin families. Twin Res. Hum. Genet. 2006, 9, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Seppälä, I.; Oksala, N.; Lyytikäinen, L.P.; Raitakari, O.; Viikari, J.; Ala-Korpela, M.; Soininen, P.; Kangas, A.J.; Waldenberger, M.; et al. Blood microRNA profile associates with the levels of serum lipids and metabolites associated with glucose metabolism and insulin resistance and pinpoints pathways underlying metabolic syndrome: The cardiovascular risk in Young Finns Study. Mol. Cell Endocrinol. 2014, 391, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar]
- Loh, P.-R.; Kichaev, G.; Gazal, S.; Schoech, A.P.; Price, A.L. Mixed-model association for biobank-scale datasets. Nat. Genet. 2018, 50, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Soranzo, N.; Sanna, S.; Wheeler, E.; Gieger, C.; Radke, D.; Dupuis, J.; Bouatia-Naji, N.; Langenberg, C.; Prokopenko, I.; Stolerman, E.; et al. Common variants at 10 genomic loci influence hemoglobin A₁(C) levels via glycemic and nonglycemic pathways. Diabetes 2010, 59, 3229–3239. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Zhang, H.-M.; Xie, G.-Y.; Miao, Y.-R.; Xia, M.; Guo, A.-Y. hTFtarget: A Comprehensive Database for Regulations of Human Transcription Factors and Their Targets. Genom. Proteom. Bioinform. 2020, 18, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.A.; Warpeha, K.; Xu, W.; Walker, B.; Trimble, E.R. High glucose decreases intracellular glutathione concentrations and upregulates inducible nitric oxide synthase gene expression in intestinal epithelial cells. J. Mol. Endocrinol. 2004, 33, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C.; Davis, J. Review: Targeting therapeutics against glutathione depletion in diabetes and its complications. Br. J. Diabetes Vasc. Dis. 2007, 7, 258–265. [Google Scholar] [CrossRef]
- Bagherion, M.; Qujeq, D.; Bakhshi Khaniniki, G.R. Evaluation of Serum Uric Acid and Glutathione Levels in Diabetic Patients and Healthy Subjects. IJDO 2013, 5, 157–162. [Google Scholar]
- Zhang, J.; An, H.; Ni, K.; Chen, B.; Li, H.; Li, Y.; Sheng, G.; Zhou, C.; Xie, M.; Chen, S.; et al. Glutathione prevents chronic oscillating glucose intake-induced β-cell dedifferentiation and failure. Cell Death Dis. 2019, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Ha, M.-H.; Kim, J.-H.; Christiani, D.C.; Gross, M.D.; Steffes, M.; Blomhoff, R.; Jacobs, D.R. Gamma-glutamyltransferase and diabetes—a 4 year follow-up study. Diabetologia 2003, 46, 359–364. [Google Scholar] [CrossRef]
- Nakanishi, N.; Suzuki, K.; Tatara, K. Serum -Glutamyltransferase and Risk of Metabolic Syndrome and Type 2 Diabetes in Middle-Aged Japanese Men. Diabetes Care 2004, 27, 1427–1432. [Google Scholar] [CrossRef]
- Sabanayagam, C.; Shankar, A.; Li, J.; Pollard, C.; Ducatman, A. Serum gamma-glutamyl transferase level and diabetes mellitus among US adults. Eur. J. Epidemiol. 2009, 24, 369–373. [Google Scholar] [CrossRef]
- Haghighi, S.; Amini, M.; Pournaghshband, Z.; Amini, P.; Hovsepian, S. Relationship between gamma-glutamyl transferase and glucose intolerance in first degree relatives of type 2 diabetics patients. J. Res. Med. Sci. 2011, 16, 123–129. [Google Scholar]
- Lee, S.-H.; Kim, K.-M.; Kim, K.-N. Combined effect of serum gamma-glutamyltransferase and uric acid on incidence of diabetes mellitus: A longitudinal study. Medicine 2017, 96, e6901. [Google Scholar] [CrossRef]
- Zhao, W.; Tong, J.; Liu, J.; Liu, J.; Li, J.; Cao, Y. The Dose-Response Relationship between Gamma-Glutamyl Transferase and Risk of Diabetes Mellitus Using Publicly Available Data: A Longitudinal Study in Japan. Int. J. Endocrinol. 2020, 2020, 5356498. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.B. Gamma Glutamyl Transferase. Crit. Rev. Clin. Lab. Sci. 2001, 38, 263–355. [Google Scholar] [CrossRef]
- Noordam, R.; Smit, R.A.; Postmus, I.; Trompet, S.; Van Heemst, D. Assessment of causality between serum gamma-glutamyltransferase and type 2 diabetes mellitus using publicly available data: A Mendelian randomization study. Int. J. Epidemiol. 2016, 45, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Nano, J.; Muka, T.; Ligthart, S.; Hofman, A.; Murad, S.D.; Janssen, H.L.A.; Franco, O.; Dehghan, A. Gamma-glutamyltransferase levels, prediabetes and type 2 diabetes: A Mendelian randomization study. Int. J. Epidemiol. 2017, 46, 1400–1409. [Google Scholar] [CrossRef]
- Murakami, K.; Takahito, K.; Ohtsuka, Y.; Fujiwara, Y.; Shimada, M.; Kawakami, Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism 1989, 38, 753–758. [Google Scholar] [CrossRef]
- Forrester, T.E.; Badaloo, V.; Bennett, F.I.; Jackson, A.A. Excessive excretion of 5-oxoproline and decreased levels of blood glutathione in type II diabetes mellitus. Eur. J. Clin. Nutr. 1990, 44, 847–850. [Google Scholar]
- Yoshida, K.; Hirokawa, J.; Tagami, S.; Kawakami, Y.; Urata, Y.; Kondo, T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: Regulation of glutathione synthesis and efflux. Diabetologia 1995, 38, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Samiec, P.S.; Drews-Botsch, C.; Flagg, E.W.; Kurtz, J.C.; Sternberg, P.; Reed, R.L.; Jones, D.P. Glutathione in Human Plasma: Decline in Association with Aging, Age-Related Macular Degeneration, and Diabetes. Free Radic. Biol. Med. 1998, 24, 699–704. [Google Scholar] [CrossRef]
- Khamaisi, M.; Cavel, O.; Rosenstock, M.; Porat, M.; Yuli, M.; Kaiser, N.; Rudich, A. Effect of inhibition of glutathione synthesis on insulin action: In vivo and in vitro studies using buthionine sulfoximine. Biochem. J. 2000, 349, 579–586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, Y.; Tran, P.O.; Harmon, J.; Robertson, R.P. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar] [CrossRef]
- Githens, S. Glutathione metabolism in the pancreas compared with that in the liver, kidney, and small intestine. Int. J. Pancreatol. 1991, 8, 97–109. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Presti, M.E.; Wells, L.D. Glutathione Synthesis in the Exocrine Pancreas. Pancreas 1997, 14, 342–349. [Google Scholar] [CrossRef]
- Wallig, M.A. Xenobiotic metabolism, oxidant stress and chronic pancreatitis. Focus on glutathione. Digestion 1998, 59 (Suppl. 4), 13–24. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.M.; Hysenaj, G.; Wood, A.R.; Weedon, M.N.; Harries, L.W. Targeted Allelic Expression Profiling in Human Islets Identifiescis-Regulatory Effects for Multiple Variants Identified by Type 2 Diabetes Genome-Wide Association Studies. Diabetes 2014, 64, 1484–1491. [Google Scholar] [CrossRef]
- Ding, L.; Fan, L.; Xu, X.; Fu, J.; Xue, Y. Identification of core genes and pathways in type 2 diabetes mellitus by bioinformatics analysis. Mol. Med. Rep. 2019, 20, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Njålsson, R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. 2005, 62, 1938–1945. [Google Scholar] [CrossRef]
- Atwal, P.S.; Medina, C.R.; Burrage, L.C.; Sutton, V.R. Nineteen-year follow-up of a patient with severe glutathione synthetase deficiency. J. Hum. Genet. 2016, 61, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; De Marco, G.; Furriol, J.; Mansego, M.; Pineda-Alonso, M.; Gonzalez-Neira, A.; Martin-Escudero, J.C.; Benitez, J.; Lluch, A.; Chaves, F.J.; et al. Oxidative stress in susceptibility to breast cancer: Study in Spanish population. BMC Cancer 2014, 14, 861. [Google Scholar] [CrossRef] [PubMed]
- Koutros, S.; Andreotti, G.; Berndt, S.I.; Barry, K.H.; Lubin, J.H.; Hoppin, J.; Kamel, F.; Sandler, D.P.; Burdette, L.A.; Yuenger, J.; et al. Xenobiotic-metabolizing gene variants, pesticide use, and the risk of prostate cancer. Pharmacogenet. Genom. 2011, 21, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Foroud, T.; Hinrichs, A.L.; Le, N.X.; Bertelsen, S.; Budde, J.P.; Harari, O.; Koller, D.L.; Wetherill, L.; Agrawal, A.; et al. A genome-wide association study of alcohol-dependence symptom counts in extended pedigrees identifies C15orf53. Mol. Psychiatry 2012, 18, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Oudot-Mellakh, T.; Cohen, W.; Germain, M.; Saut, N.; Kallel, C.; Zelenika, D.; Lathrop, M.; Trégouët, D.-A.; Morange, P.-E. Genome wide association study for plasma levels of natural anticoagulant inhibitors and protein C anticoagulant pathway: The MARTHA project. Br. J. Haematol. 2012, 157, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- Hellwege, J.N.; Edwards, D.R.V.; Giri, A.; Qiu, C.; Park, J.; Torstenson, E.S.; Keaton, J.M.; Wilson, O.D.; Robinson-Cohen, C.; Chung, C.P.; et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2018, 104, 65–75. [Google Scholar] [CrossRef]
- Tang, W.; Basu, S.; Kong, X.; Pankow, J.S.; Aleksic, N.; Tan, A.; Cushman, M.; Boerwinkle, E.; Folsom, A.R. Genome-wide association study identifies novel loci for plasma levels of protein C: The ARIC study. Blood 2010, 116, 5032–5036. [Google Scholar] [CrossRef]
- Njälsson, R.; Norgren, S. Physiological and pathological aspects of GSH metabolism. Acta Paediatr. 2007, 94, 132–137. [Google Scholar] [CrossRef]
- Fawaz, M.V.; Topper, M.E.; Firestine, S.M. The ATP-grasp enzymes. Bioorganic Chem. 2011, 39, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Sant, K.; Hansen, J.M.; Williams, L.; Tran, N.L.; Goldstone, J.; Stegeman, J.J.; Hahn, M.; Timme-Laragy, A. The role of Nrf1 and Nrf2 in the regulation of glutathione and redox dynamics in the developing zebrafish embryo. Redox Biol. 2017, 13, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Blomhoff, R.; Jacobs, D.R. ReviewIs Serum Gamma Glutamyltransferase a Marker of Oxidative Stress? Free Radic. Res. 2004, 38, 535–539. [Google Scholar] [CrossRef] [PubMed]
- McConnachie, L.A.; Mohar, I.; Hudson, F.N.; Ware, C.B.; Ladiges, W.C.; Fernandez, C.; Chatterton-Kirchmeier, S.; White, C.C.; Pierce, R.H.; Kavanagh, T.J. Glutamate Cysteine Ligase Modifier Subunit Deficiency and Gender as Determinants of Acetaminophen-Induced Hepatotoxicity in Mice. Toxicol. Sci. 2007, 99, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Hamon, I.; Valdes, V.; Franck, P.; Buchweiller, M.C.; Fresson, J.; Hascoet, J.M. Gender-dependent differences in glutathione (GSH) metabolism in very preterm infants. Arch. Pediatr. 2011, 18, 247–252. (In French) [Google Scholar] [CrossRef] [PubMed]
- Massafra, C.; Gioia, D.; De Felice, C.; Muscettola, M.; Longini, M.; Buonocore, G. Gender-related differences in erythrocyte glutathione peroxidase activity in healthy subjects. Clin. Endocrinol. 2002, 57, 663–667. [Google Scholar] [CrossRef]
- Das, M.; Dixit, R.; Seth, P.K.; Mukhtar, H. Glutathione-S-Transferase Activity in the Brain: Species, Sex, Regional, and Age Differences. J. Neurochem. 1981, 36, 1439–1442. [Google Scholar] [CrossRef]
- Wang, L.; Ahn, Y.J.; Asmis, R. Sexual dimorphism in glutathione metabolism and glutathione-dependent responses. Redox Biol. 2019, 31, 101410. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Dröge, W.; Breitkreutz, R. Glutathione and immune function. Proc. Nutr. Soc. 2000, 59, 595–600. [Google Scholar] [CrossRef]
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties. Front. Immunol. 2017, 8, 1239. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Asp. Med. 2009, 30, 77–85. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Bulleid, N. Glutathione Is Required to Regulate the Formation of Native Disulfide Bonds within Proteins Entering the Secretory Pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef] [PubMed]
- Mytilineou, C.; Kramer, B.C.; Yabut, J.A. Glutathione depletion and oxidative stress. Park. Relat. Disord. 2002, 8, 385–387. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Sheena, Y.; Land, J.M.; Heales, S.J.R. Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2005, 28, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar] [CrossRef]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Melo, E.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. eLife 2014, 3, e03421. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.P.; Wetlaufer, D.B. Formation of three-dimensional structure in proteins. I. Rapid nonenzymic reactivation of reduced lysozyme. Biochemistry 1970, 9, 5015–5023. [Google Scholar] [CrossRef] [PubMed]
- Creighton, T.E. Intermediates in the refolding of reduced ribonuclease A. J. Mol. Biol. 1979, 129, 411–431. [Google Scholar] [CrossRef]
- Arolas, J.L.; Aviles, F.X.; Chang, J.-Y.; Ventura, S. Folding of small disulfide-rich proteins: Clarifying the puzzle. Trends Biochem. Sci. 2006, 31, 292–301. [Google Scholar] [CrossRef]
- Okumura, M.; Saiki, M.; Yamaguchi, H.; Hidaka, Y. Acceleration of disulfide-coupled protein folding using glutathione derivatives. FEBS J. 2011, 278, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.; Lodish, H. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Konishi, Y.; Ooi, T.; Scheraga, H.A. Regeneration of ribonuclease A from the reduced protein. 1. Rate-limiting steps. Biochemistry 1982, 21, 4734–4740. [Google Scholar] [CrossRef]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Dependence of the rate on the composition of the redox buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards, G., III; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20, 28–50. [Google Scholar] [CrossRef]
- Scheuner, D.; Kaufman, R.J. The Unfolded Protein Response: A Pathway That Links Insulin Demand with β-Cell Failure and Diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Nyamdorj, R. Is the association of type II diabetes with waist circumference or waist-to-hip ratio stronger than that with body mass index? Eur. J. Clin. Nutr. 2009, 64, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Wittemans, L.B.L.; Zuber, V.; Stewart, I.D.; Sharp, S.J.; Luan, J.; Day, F.R.; Li, C.; Bowker, N.; Cai, L.; et al. Association of Genetic Variants Related to Gluteofemoral vs Abdominal Fat Distribution with Type 2 Diabetes, Coronary Disease, and Cardiovascular Risk Factors. JAMA 2018, 320, 2553–2563. [Google Scholar] [CrossRef]
- Graff, M.; Scott, R.A.; Justice, A.E.; Young, K.L.; Feitosa, M.F.; Barata, L.; Winkler, T.W.; Chu, A.Y.; Mahajan, A.; Hadley, D.; et al. Genome-wide physical activity interactions in adiposity—A meta-analysis of 200,452 adults. PLoS Genet. 2017, 13, e1006528. [Google Scholar] [CrossRef]
- Mohan Rao, L.V.; Esmon, C.T.; Pendurthi, U.R. Endothelial cell protein C receptor: A multiliganded and multifunctional receptor. Blood 2014, 124, 1553–1562. [Google Scholar] [CrossRef]
- Lattenist, L.; Ochodnický, P.; Ahdi, M.; Claessen, N.; Leemans, J.C.; Satchell, S.C.; Florquin, S.; Gerdes, V.E.; Roelofs, J.J.T.H. Renal endothelial protein C receptor expression and shedding in diabetic nephropathy. J. Thromb. Haemost. 2016, 14, 1171–1182. [Google Scholar] [CrossRef]
- Isermann, B.; Vinnikov, I.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef]
- Contreras, J.L.; Eckstein, C.; Smyth, C.A.; Bilbao, G.; Vilatoba, M.; Ringland, S.E.; Young, C.; Thompson, J.A.; Fernandez, J.; Griffin, J.H.; et al. Activated Protein C Preserves Functional Islet Mass After Intraportal Transplantation: A Novel Link Between Endothelial Cell Activation, Thrombosis, Inflammation, and Islet Cell Death. Diabetes 2004, 53, 2804–2814. [Google Scholar] [CrossRef]
- Hong, Y.; Ohishi, K.; Kang, J.Y.; Tanaka, S.; Inoue, N.; Nishimura, J.-I.; Maeda, Y.; Kinoshita, T. Human PIG-U and Yeast Cdc91p Are the Fifth Subunit of GPI Transamidase That Attaches GPI-Anchors to Proteins. Mol. Biol. Cell 2003, 14, 1780–1789. [Google Scholar] [CrossRef]
- Marchese, A.; Raiborg, C.; Santini, F.; Keen, J.H.; Stenmark, H.; Benovic, J.L. The E3 Ubiquitin Ligase AIP4 Mediates Ubiquitination and Sorting of the G Protein-Coupled Receptor CXCR4. Dev. Cell 2003, 5, 709–722. [Google Scholar] [CrossRef]
- Shembade, N.; Parvatiyar, K.; Harhaj, N.S.; Harhaj, E.W. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-κB signalling. EMBO J. 2009, 28, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, C.; Gao, K.; Wang, D.; Mao, J.; An, J.; Xu, C.; Wu, D.; Yu, H.; Liu, J.O.; et al. The Ubiquitin Ligase Itch Regulates Apoptosis by Targeting Thioredoxin-interacting Protein for Ubiquitin-dependent Degradation. J. Biol. Chem. 2010, 285, 8869–8879. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Martínez-Micaelo, N.; Gonzalez-Abuin, N.; Terra, X.; Ardévol, A.; Pinent, M.; Petretto, E.; Behmoaras, J.; Blay, M.T. Identification of a nutrient sensing transcriptional network in monocytes by using inbred rat models of cafeteria diet. Dis. Model. Mech. 2016, 9, 1231–1239. [Google Scholar] [CrossRef]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic Depletion of the Energy Metabolite Acetyl-Coenzyme A Stimulates Autophagy and Prolongs Lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef]
- Dill, K.A.; Maccallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Langen, R.; Gurlo, T.; Matveyenko, A.V.; Butler, P.C. β-Cell Failure in Type 2 Diabetes: A Case of Asking Too Much of Too Few? Diabetes 2013, 62, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Allard, C.; Alvarez-Mercado, A.I.; Fuselier, T.; Kim, J.H.; Coons, L.; Hewitt, S.; Urano, F.; Korach, K.S.; Levin, E.R.; et al. Estrogens Promote Misfolded Proinsulin Degradation to Protect Insulin Production and Delay Diabetes. Cell Rep. 2018, 24, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Aspects Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Manickam, N.; Soliman, A.; Tsai, B.; Liu, M.; Arvan, P. Disulfide Mispairing During Proinsulin Folding in the Endoplasmic Reticulum. Diabetes 2016, 65, 1050–1060. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 11, e44532. [Google Scholar] [CrossRef]
- Leyva-García, E.; Lara-Martínez, R.; Morán-Zanabria, L.; Monsalve, M.C.R.; Jiménez-García, L.F.; Oviedo, N.; Murata, C.; Garrido-Magaña, E.; Altamirano-Bustamante, N.F.; Altamirano-Bustamante, M.M. Novel insight into streptozotocin-induced diabetic rats from the protein misfolding perspective. Sci. Rep. 2017, 7, 11552. [Google Scholar] [CrossRef]
- Wang, Z.; Gurlo, T.; Matveyenko, A.V.; Elashoff, D.; Wang, P.; Rosenberger, M.; Junge, J.A.; Stevens, R.C.; White, K.L.; Fraser, S.E.; et al. Live-cell imaging of glucose-induced metabolic coupling of β and α cell metabolism in health and type 2 diabetes. Commun. Biol. 2021, 4, 594. [Google Scholar] [CrossRef]
- Ueno, Y.; Kizaki, M.; Nakagiri, R.; Kamiya, T.; Sumi, H.; Osawa, T. Dietary Glutathione Protects Rats from Diabetic Nephropathy and Neuropathy. J. Nutr. 2002, 132, 897–900. [Google Scholar] [CrossRef]
- El-Hafidi, M.; Franco, M.; Ramírez, A.R.; Sosa, J.S.; Flores, J.A.P.; Acosta, O.L.; Salgado, M.C.; Cardoso-Saldaña, G. Glycine Increases Insulin Sensitivity and Glutathione Biosynthesis and Protects against Oxidative Stress in a Model of Sucrose-Induced Insulin Resistance. Oxid. Med. Cell. Longev. 2018, 2018, 2101562. [Google Scholar] [CrossRef]
- Lapenna, D.; De Gioia, S.; Ciofani, G.; Mezzetti, A.; Ucchino, S.; Calafiore, A.M.; Napolitano, A.M.; Di Ilio, C.; Cuccurullo, F. Glutathione-Related Antioxidant Defenses in Human Atherosclerotic Plaques. Circulation 1998, 97, 1930–1934. [Google Scholar] [CrossRef]
- Robaczewska, J.; Kedziora-Kornatowska, K.; Kozakiewicz, M.; Zary-Sikorska, E.; Pawluk, H.; Pawliszak, W.; Kedziora, J. Role of glutathione metabolism and glutathione-related antioxidant defense systems in hypertension. J. Physiol. Pharmacol. 2016, 67, 331–337. [Google Scholar] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Vairetti, M.; Di Pasqua, L.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef]
- Mannino, G.C.; Andreozzi, F.; Sesti, G. Pharmacogenetics of type 2 diabetes mellitus, the route toward tailored medicine. Diabetes Metab. Res. Rev. 2018, 35, e3109. [Google Scholar] [CrossRef]
- Chernikova, N.A.; Kamynina, L.L.; Ametov, A.S.; Sychev, D.A.; Grishina, E.A.; Ryzhikova, K.A. The pharmacogenetics of hypoglycemia and the glycemic variability at the patients ith type 2 diabetes mellitus. Terapevticheskii Arkhiv 2020, 92, 54–62. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | Healthy Controls, n = 1626 | T2D Patients, n = 1572 | p-Value | |
|---|---|---|---|---|
| Age, mean ± standard deviation | 60.8 ± 6.4 | 61.3 ± 10.4 | 0.34 | |
| Sex | Males, n (%) | 601 (37.0) | 584 (37.2) | 0.89 |
| Females, n (%) | 1024 (63.0) | 988 (62.8) | ||
| Body mass index (kg/m2), mean ± standard deviation | 27.21 ± 3.55 | 31.92 ± 6.65 | 0.001 | |
| Duration of diabetes, median (Q1; Q3) | - | 9.0 (3.0; 15.0) | - | |
| Hypertension, n (%) | 24 (1.5) | 1340 (85.2) | <0.0001 | |
| CAD, n (%) | - | 511 (32.5) | <0.0001 | |
| Smokers (ever/never), n (%) | 501 (31.0%) | 412 (26.1%) | 0.004 | |
| Positive family history of diabetes, n (%) | 33 (2.1%) | 602 (38.1%) | <0.0001 | |
| HbA1C (%), Me (Q1; Q3) | 4.58 (4.11; 4.87) | 9.02 (7.70; 10.80) | <0.0001 | |
| FBG (mmol/L), Me (Q1; Q3) | 4.71 (4.39; 4.84) | 12.20 (9.70; 15.20) | <0.0001 | |
| Total cholesterol (mmol/L), Me (Q1; Q3) | 3.06 (2.86; 3.12) | 5.10 (4.27; 6.09) | <0.0001 | |
| LDL (mmol/L), Me (Q1; Q3) | 1.74 (1.60; 1.79) | 3.03 (2.40; 4.05) | <0.0001 | |
| HDL (mmol/L), Me (Q1; Q3) | 1.47 (1.36; 1.62) | 0.85 (0.74; 1.07) | <0.0001 | |
| Triacylglycerides (mmol/L), Me (Q1; Q3) | 1.15 (0.98; 1.23) | 2.20 (1.55; 3.00) | <0.0001 | |
| H2O2 (mmol/L), Me(Q1;Q3) | 2.81 (2.18; 3.63) | 3.70 (2.65; 4.97) | <0.0001 | |
| GSSG/GSH (mmol/L), Me(Q1;Q3) | 1.91 (0.83; 5.41) | 1.63 (0.56; 3.79) | 0.037 | |
| Gene, SNP | Genotype, Allele | Healthy Controls, n = 1626 n (%) 1 | T2D Patients, n = 1572 n (%) 1 | OR (95% CI) 2 | p-Value 2 | adjOR (95% CI) 3 | p-Value 3 |
|---|---|---|---|---|---|---|---|
| GSS rs13041792 G > A | G/G | 1022 (66) | 957 (61.9) | 1.00 | 0.046 | 1.00 | 0.027 |
| G/A | 460 (29.7) | 523 (33.8) | 1.21 (1.04–1.42) | 1.24 (1.06–1.44) | |||
| A/A | 66 (4.3) | 66 (4.3) | 1.07 (0.75–1.52) | 1.07 (0.75–1.53) | |||
| A | 0.191 | 0.212 | 1.14 (1.01–1.29) | 0.043 | - | 0.05 | |
| GSS rs1801310 G > A | G/G | 590 (36.4) | 581 (36.8) | 1.00 | 0.16 | 1.00 | 0.18 |
| G/A | 761 (47) | 773 (49) | 1.03 (0.89–1.20) | 1.04 (0.89–1.21) | |||
| A/A | 270 (16.7) | 225 (14.2) | 0.85 (0.69–1.04) | 0.85 (0.69–1.06) | |||
| A | 0.401 | 0.387 | 0.94 (0.85–1.04) | 0.25 | - | 0.38 | |
| GSS rs6088660 C > T | C/C | 792 (50.5) | 815 (52.4) | 1.00 | 0.44 | 1.00 | 0.43 |
| C/T | 647 (41.3) | 628 (40.4) | 0.94 (0.81–1.09) | 0.94 (0.81–1.09) | |||
| T/T | 128 (8.2) | 112 (7.2) | 0.85 (0.65–1.12) | 0.85 (0.64–1.12) | |||
| T | 0.288 | 0.274 | 0.93 (0.83–1.04) | 0.21 | - | 0.26 | |
| GGT7 rs11546155 G > A | G/G | 1297 (80.5) | 1283 (81.3) | 1.00 | 0.013 | 1.00 | 0.022 |
| G/A | 282 (17.5) | 282 (17.9) | 1.01 (0.84–1.21) | 1.02 (0.85–1.22) | |||
| A/A | 33 (2) | 13 (0.8) | 0.40 (0.21–0.76) | 0.42 (0.22–0.80) | |||
| A | 0.108 | 0.098 | 0.89 (0.76–1.05) | 0.17 | 0.21 | ||
| GGT7 rs6119534 C > T | C/C | 241 (19.8) | 352 (27.1) | 1.00 | 0.0001 | 1.00 | 0.0003 |
| C/T | 864 (71) | 832 (64.2) | 0.63 (0.51–0.79) | 0.67 (0.56–0.82) | |||
| T/T | 111 (9.1) | 113 (8.7) | 0.62 (0.43–0.88) | 0.73 (0.53–1.00) | |||
| T | 0.447 | 0.408 | 0.85 (0.76–0.95) | 0.006 | - | 0.012 |
| Haplotypes of the GSS Gene (Global Haplotype Association p-Value 0.039) | |||||||
| rs1801310 | rs6088660 | rs13041792 | Healthy Controls (n = 1626) | T2D Patients (n = 1572) | OR (95% CI) * | p-Value (Q-Value) | |
| A | C | G | 0.3915 | 0.3782 | 1.00 | - | |
| G | T | G | 0.2823 | 0.2713 | 1.02 (0.89–1.17) | 0.78 (0.91) | |
| G | C | A | 0.1821 | 0.203 | 1.26 (1.08–1.47) | 0.003 (0.011) | |
| G | C | G | 0.1334 | 0.1373 | 0.99 (0.83–1.18) | 0.92 (0.92) | |
| Haplotypes of the GGT7 Gene (Global Haplotype Association p-Value 0.0018) | |||||||
| rs6119534 | rs11546155 | Healthy Controls (n = 1626) | T2D patients (n = 1572) | OR (95% CI) * | p-value (Q-value) | ||
| C | G | 0.4570 | 0.4992 | 1.00 | - | ||
| T | G | 0.4350 | 0.4032 | 0.80 (0.68–0.95) | 0.009 (0.026) | ||
| C | A | 0.0971 | 0.0931 | 0.76 (0.61–0.94) | 0.011 (0.026) | ||
| SNP–SNP Interaction mbmdr-Models | NH | β H | WH | NL | β L | WL | pperm | |
|---|---|---|---|---|---|---|---|---|
| 1 | GGT7 rs6119534 × GSS rs13041792 | 2 | 0.111 | 30.53 | 1 | −0.118 | 32.37 | <0.001 |
| 2 | GGT7 rs6119534 × GSS rs1801310 | 2 | 0.104 | 17.66 | 1 | −0.256 | 22.74 | <0.001 |
| 3 | GGT7 rs11546155 × GGT7 rs6119534 | 2 | 0.109 | 21.07 | 3 | −0.079 | 13.93 | <0.001 |
| 4 | GGT7 rs6119534 × GSS rs6088660 | 2 | 0.107 | 20.08 | 2 | −0.060 | 8.36 | <0.001 |
| 5 | GGT7 rs11546155 × GSS rs6088660 | 0 | NA | NA | 1 | −0.319 | 8.93 | 0.034 |
| 6 | GGT7 rs11546155 × GSS rs1801310 | 0 | NA | NA | 1 | −0.223 | 7.89 | 0.046 |
| 7 | GGT7 rs11546155 × GSS rs13041792 | 1 | 0.040 | 4.05 | 1 | −0.208 | 7.52 | 0.046 |
| No. | Genotype Combination | T2D Patients | Healthy Controls | OR (95% CI) 1 | p2 | Q3 | ||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | |||||
| G1 | GGT7 rs6119534-C/C × GSS rs13041792-G/G | 333 | 26.1 | 223 | 18.9 | 1.52 (1.25–1.84) | 0.00002 | 0.0001 |
| G2 | GGT7 rs6119534-C/T × GSS rs13041792-G/G | 409 | 32.1 | 509 | 43.1 | 0.62 (0.53–0.73) | 1.6 × 10−8 | 2.9 × 10−7 |
| G3 | GGT7 rs6119534-C/T × GSS rs13041792-G/A | 379 | 29.7 | 305 | 25.8 | 1.21 (1.02–1.45) | 0.03 | 0.04 |
| G4 | GGT7 rs6119534-C/C × GSS rs1801310-G/A | 168 | 13.0 | 109 | 9.0 | 1.51 (1.17–1.94) | 0.002 | 0.01 |
| G5 | GGT7 rs6119534-C/C × GSS rs1801310-A/A | 135 | 10.4 | 93 | 7.7 | 1.40 (1.06–1.84) | 0.017 | 0.03 |
| G6 | GGT7 rs6119534-C/T × GSS rs1801310-A/A | 24 | 1.9 | 65 | 5.4 | 0.33 (0.21–0.54) | 2.0 × 10−6 | 1.8 × 10−5 |
| G7 | GGT7 rs6119534-C/C × GGT7 rs11546155-G/G | 247 | 19.0 | 163 | 13.4 | 1.52 (1.22–1.88) | 0.0001 | 0.0005 |
| G8 | GGT7 rs6119534-C/C × GGT7 rs11546155-G/A | 98 | 7.6 | 67 | 5.5 | 1.40 (1.02–1.93) | 0.04 | 0.042 |
| G9 | GGT7 rs6119534-C/T × GGT7 rs11546155-G/G | 696 | 53.7 | 703 | 57.9 | 0.84 (0.72–0.99) | 0.03 | 0.04 |
| G10 | GGT7 rs6119534-C/T × GGT7 rs11546155-A/A | 1 | 0.1 | 9 | 0.7 | 0.15 (0.03–0.82) | 0.02 | 0.03 |
| G11 | GGT7 rs6119534-C/C × GSS rs6088660-C/C | 236 | 18.3 | 156 | 13.0 | 1.50 (1.21–1.87) | 0.0003 | 0.001 |
| G12 | GGT7 rs6119534-C/C × GSS rs6088660-C/T | 103 | 8.0 | 70 | 5.8 | 1.40 (1.03–1.92) | 0.03 | 0.04 |
| G13 | GGT7 rs6119534-C/T × GSS rs6088660-C/T | 367 | 28.5 | 388 | 32.3 | 0.83 (0.70–0.99) | 0.04 | 0.042 |
| G14 | GGT7 rs6119534-C/T × GSS rs6088660-T/T | 56 | 4.3 | 73 | 6.1 | 0.70 (0.49–1.00) | 0.05 | 0.05 |
| G15 | GGT7 rs11546155-A/A × GSS rs6088660-C/T | 4 | 0.3 | 18 | 1.2 | 0.24 (0.09–0.68) | 0.01 | 0.02 |
| G16 | GGT7 rs11546155-A/A × GSS rs1801310-G/G | 11 | 0.7 | 29 | 1.8 | 0.38 (0.19–0.77) | 0.005 | 0.01 |
| G17 | GGT7 rs11546155-G/G × GSS rs13041792-G/A | 457 | 29.6 | 406 | 26.3 | 1.18 (1.00–1.38) | 0.04 | 0.042 |
| G18 | GGT7 rs11546155-A/A × GSS rs13041792-G/G | 13 | 0.8 | 31 | 2.0 | 0.41 (0.22–0.79) | 0.006 | 0.01 |
| Gene, SNP | Genotype | Entire Group | Males | Females | |||
|---|---|---|---|---|---|---|---|
| T2D Patients (n = 489) | p-Value * (Q-Value) | T2D Patients (n = 145) | p-Value * (Q-Value) | T2D Patients (n = 344) | p-Value * (Q-Value) | ||
| H2O2, µmol/L | |||||||
| GSS rs13041792 G > A | G/G | 3.67 (2.66; 4.96) | 0.54 (0.72) | 3.55 (2.44; 4.81) | 0.17 (0.31) | 3.79 (2.73; 5.02) | 0.56 (0.72) |
| G/A | 3.77 (2.65; 5.09) | 3.46 (2.39; 4.47) | 3.80 (2.74; 5.10) | ||||
| A/A | 3.88 (2.76; 5.44) | 3.67 (2.61; 6.94) | 3.88 (2.76; 5.38) | ||||
| GSS rs1801310 G > A | G/G | 3.56 (2.64; 4.91) | 0.56 (0.72) | 3.26 (2.29; 4.07) | 0.26 (0.42) | 3.75 (2.70; 5.04) | 0.88 (0.90) |
| G/A | 3.80 (2.72; 5.14) | 3.59 (2.42; 4.81) | 3.88 (2.90; 5.17) | ||||
| A/A | 3.68 (2.46; 4.61) | 4.37 (2.46; 5.33) | 3.63 (2.49; 4.22) | ||||
| GSS rs6088660 C > T | C/C | 3.77 (2.54; 4.96) | 0.056 (0.18) | 3.66 (2.30; 5.13) | 0.73 (0.78) | 3.81 (2.70; 4.94) | 0.039 (0.18) |
| C/T | 3.79 (2.83; 5.21) | 3.76 (2.60; 5.18) | 3.79 (2.90; 5.23) | ||||
| T/T | 3.37 (2.64; 4.07) | 3.21 (2.41; 3.56) | 3.43 (2.64; 5.87) | ||||
| GGT7 rs11546155 G > A | G/G | 3.67 (2.63; 4.96) | 0.62 (0.73) | 3.55 (2.42; 4.81) | 0.30 (0.45) | 3.72 (2.66; 4.96) | 0.046 (0.18) |
| G/A | 3.95 (2.74; 5.12) | 3.01 (2.20; 4.24) | 4.48 (3.23; 5.35) | ||||
| A/A | 3.67 (2.64; 5.04) | 2.08 (2.08; 2.08) | 4.15 (3.19; 5.87) | ||||
| GGT7rs6119534 C > T | C/C | 3.33 (2.20; 4.42) | 0.0002 (0.009) | 3.10 (2.13; 4.24) | 0.05 (0.18) | 3.39 (2.22; 4.82) | 0.0009 (0.02) |
| C/T | 3.74 (2.76; 5.05) | 3.60 (2.42; 4.79) | 3.79 (2.90; 5.09) | ||||
| T/T | 3.97 (3.05; 7.93) | 3.61 (3.08; 7.93) | 4.63 (2.76; 7.74) | ||||
| Total glutathione, µmol/L | |||||||
| GSS rs13041792 G > A | G/G | 1.41 (0.60; 3.67) | 0.52 (0.72) | 2.11 (0.91; 3.86) | 0.60 (0.73) | 1.34 (0.56; 3.53) | 0.26 (0.42) |
| G/A | 2.31 (0.46; 4.01) | 1.40 (0.67; 3.82) | 2.42 (0.44; 4.03) | ||||
| A/A | 0.84 (0.27; 3.65) | 2.56 (1.02; 4.23) | 0.64 (0.27; 1.35) | ||||
| GSS rs1801310 G > A | G/G | 2.43 (0.54; 3.92) | 0.26 (0.42) | 2.16 (0.77; 3.84) | 0.67 (0.75) | 2.44 (0.46; 4.07) | 0.039 (0.18) |
| G/A | 1.40 (0.58; 3.68) | 1.64 (0.74; 3.75) | 1.28 (0.48; 3.53) | ||||
| A/A | 1.30 (0.56; 3.18) | 1.78 (0.60; 3.94) | 1.29 (0.56; 2.70) | ||||
| GSS rs6088660 C > T | C/C | 1.35 (0.56; 3.65) | 0.16 (0.30) | 2.20 (0.88; 3.92) | 0.70 (0.77) | 1.30 (0.49; 3.35) | 0.086 (0.19) |
| C/T | 2.01 (0.53; 3.88) | 1.31 (0.50; 3.67) | 2.28 (0.54; 3.89) | ||||
| T/T | 2.11 (0.77; 4.21) | 2.12 (1.23; 3.86) | 2.93 (0.40; 4.36) | ||||
| GGT7 rs11546155 G > A | G/G | 1.47 (0.54; 3.68) | 0.077 (0.18) | 1.47 (0.61; 3.75) | 0.023 (0.18) | 1.46 (0.51; 3.68) | 0.66 (0.75) |
| G/A | 2.01 (0.65; 4.21) | 3.55 (1.05; 4.42) | 1.49 (0.39; 4.19) | ||||
| A/A | 3.11 (1.42; 4.09) | 3.11 (2.45; 3.77) | 2.39 (0.38; 4.40) | ||||
| GGT7 rs6119534 C > T | C/C | 1.36 (0.51; 3.34) | 0.13 (0.25) | 2.57 (0.88; 3.93) | 0.053 (0.18) | 1.28 (0.49; 2.90) | 0.063 (0.18) |
| C/T | 1.70 (0.51; 3.89) | 1.55 (0.56; 3.82) | 1.93 (0.46; 3.97) | ||||
| T/T | 3.91 (0.91; 4.53) | 4.70 (4.59; 3.81) | 3.57 (0.59; 4.00) | ||||
| Fasting blood glucose, mmol/L | |||||||
| GSS rs13041792 G > A | G/G | 12.00 (9.40; 15.00) | 0.032 (0.18) | 12.20 (9.20; 15.00) | 0.026 (0.18) | 11.90 (9.40; 15.00) | 0.29 (0.45) |
| G/A | 12.50 (10.00; 15.40) | 12.50 (10.10; 15.40) | 12.45 (9.92;15.35) | ||||
| A/A | 13.00 (10.20; 16.30) | 13.50 (11.90; 17.60) | 12.20 (9.06;16.20) | ||||
| GSS rs1801310 G > A | G/G | 12.40 (9.50; 15.60) | 0.47 (0.68) | 12.50 (9.16; 15.20) | 0.10 (0.20) | 12.40 (9.57;16.00) | 0.60 (0.73) |
| G/A | 12.00 (9.80; 15.00) | 12.60 (10.00; 15.20) | 12.00 (9.63; 14.90) | ||||
| A/A | 12.20 (9.40; 15.40) | 12.50 (9.00; 15.20) | 12.00 (9.45; 15.49) | ||||
| GSS rs6088660 C > T | C/C | 12.00 (9.70; 15.20) | 0.10 (0.20) | 12.50 (10.10; 15.15) | 0.007 (0.11) | 12.00 (9.51; 15.20) | 0.78 (0.82) |
| C/T | 12.20 (9.60; 15.00) | 12.62 (9.60; 15.40) | 12.00 (9.70; 15.00) | ||||
| T/T | 11.90 (8.30; 15.50) | 10.90 (7.30; 15.00) | 12.60 (9.50; 16.20) | ||||
| GGT7 rs11546155 G > A | G/G | 12.20 (9.70; 15.30) | 0.053 (0.18) | 12.50 (9.70; 15.20) | 0.06 (0.18) | 12.10 (9.70; 15.30) | 0.069 (0.18) |
| G/A | 11.60 (9.25; 14.80) | 12.70 (9.20; 15.00) | 11.45 (9.26; 14.60) | ||||
| A/A | 15.00 (7.50; 16.30) | 13.85 (10.05; 24.95) | 15.00 (7.43; 16.30) | ||||
| GGT7 rs6119534 C > T | C/C | 11.60 (9.18; 14.75) | 0.07 (0.18) | 12.15 (10.00; 14.65) | 0.96 (0.96) | 11.30 (8.83; 14.84) | 0.035 (0.18) |
| C/T | 12.17 (9.71; 15.00) | 12.60 (9.52; 15.40) | 12.00 (9.80; 15.00) | ||||
| T/T | 12.70 (10.00; 15.92) | 13.10 (10.20; 15.20) | 12.60 (9.60; 16.00) | ||||
| Gene, SNP (Effective Allele) | Phenotype | p-Value | Beta/Odds Ratio | Sample Size |
|---|---|---|---|---|
| GSS rs13041792 (A) | Waist–hip ratio adj BMI | 0.00006 | ▼−0.0103 | 1,542,860 |
| BMI | 0.0005 | ▲0.0071 | 3,167,810 | |
| Non-proliferative diabetic retinopathy | 0.018 | ▼0.9042 | 3045 | |
| Waist–hip ratio | 0.033 | ▼−0.0056 | 2,046,830 | |
| GSS rs1801310 (G) | Insulin sensitivity | 0.007 | ▼−0.0738 | 2765 |
| Insulin sensitivity adj BMI | 0.013 | ▼−0.0690 | 2764 | |
| Diabetic retinopathy | 0.014 | ▼0.9239 | 3959 | |
| Non-proliferative diabetic retinopathy | 0.018 | ▼0.9508 | 3045 | |
| Acute insulin response adj BMI–SI | 0.023 | ▼−0.0525 | 4385 | |
| Peak insulin response adj BMI–SI | 0.033 | ▼−0.0491 | 4389 | |
| Fasting plasma glucose in diabetics and non-diabetics | 0.037 | ▼−0.0191 | 16,076 | |
| Acute insulin response adj SI | 0.047 | ▼−0.0457 | 4386 | |
| GSS rs6088660 (T) | Obesity | 0.0009 | ▲1.1090 | 11,743 |
| Waist circumference adj BMI–smoking status | 0.02 | ▲0.0102 | 282,202 | |
| Fasting proinsulin | 0.04 | ▼−0.0150 | 10,701 | |
| GGT7 rs11546155 (A) | Type 2 diabetes | 0.018 | ▼0.9835 | 1,277,880 |
| Waist–hip ratio adj BMI | 0.0009 | ▲0.0077 | 1,834,510 | |
| Chronic kidney disease in type 2 diabetics | 0.004 | ▲1.2590 | 300 | |
| Fasting plasma glucose in diabetics and non-diabetics | 0.016 | ▼−0.0275 | 16,076 | |
| Neuropathy in type 2 diabetics | 0.025 | ▲1.2462 | 2344 | |
| BMI | 0.027 | ▼−0.0062 | 2,814,100 | |
| HbA1c adj BMI | 0.05 | ▲0.0496 | 7267 | |
| GGT7 rs6119534 (T) | BMI | 0.0002 | ▲0.0069 | 1,335,110 |
| Waist–hip ratio adj BMI | 0.001 | ▼−0.0063 | 971,589 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azarova, I.; Klyosova, E.; Polonikov, A. The Link between Type 2 Diabetes Mellitus and the Polymorphisms of Glutathione-Metabolizing Genes Suggests a New Hypothesis Explaining Disease Initiation and Progression. Life 2021, 11, 886. https://doi.org/10.3390/life11090886
Azarova I, Klyosova E, Polonikov A. The Link between Type 2 Diabetes Mellitus and the Polymorphisms of Glutathione-Metabolizing Genes Suggests a New Hypothesis Explaining Disease Initiation and Progression. Life. 2021; 11(9):886. https://doi.org/10.3390/life11090886
Chicago/Turabian StyleAzarova, Iuliia, Elena Klyosova, and Alexey Polonikov. 2021. "The Link between Type 2 Diabetes Mellitus and the Polymorphisms of Glutathione-Metabolizing Genes Suggests a New Hypothesis Explaining Disease Initiation and Progression" Life 11, no. 9: 886. https://doi.org/10.3390/life11090886
APA StyleAzarova, I., Klyosova, E., & Polonikov, A. (2021). The Link between Type 2 Diabetes Mellitus and the Polymorphisms of Glutathione-Metabolizing Genes Suggests a New Hypothesis Explaining Disease Initiation and Progression. Life, 11(9), 886. https://doi.org/10.3390/life11090886