
Acute and Delayed Doxorubicin-Induced Myocardiotoxicity Associated with Elevation of Cardiac Biomarkers, Depletion of Cellular Antioxidant Enzymes, and Several Histopathological and Ultrastructural Changes

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Ethical Considerations
2.1.2. Animals
2.1.3. Chemicals
2.2. Methods and Experimental Design
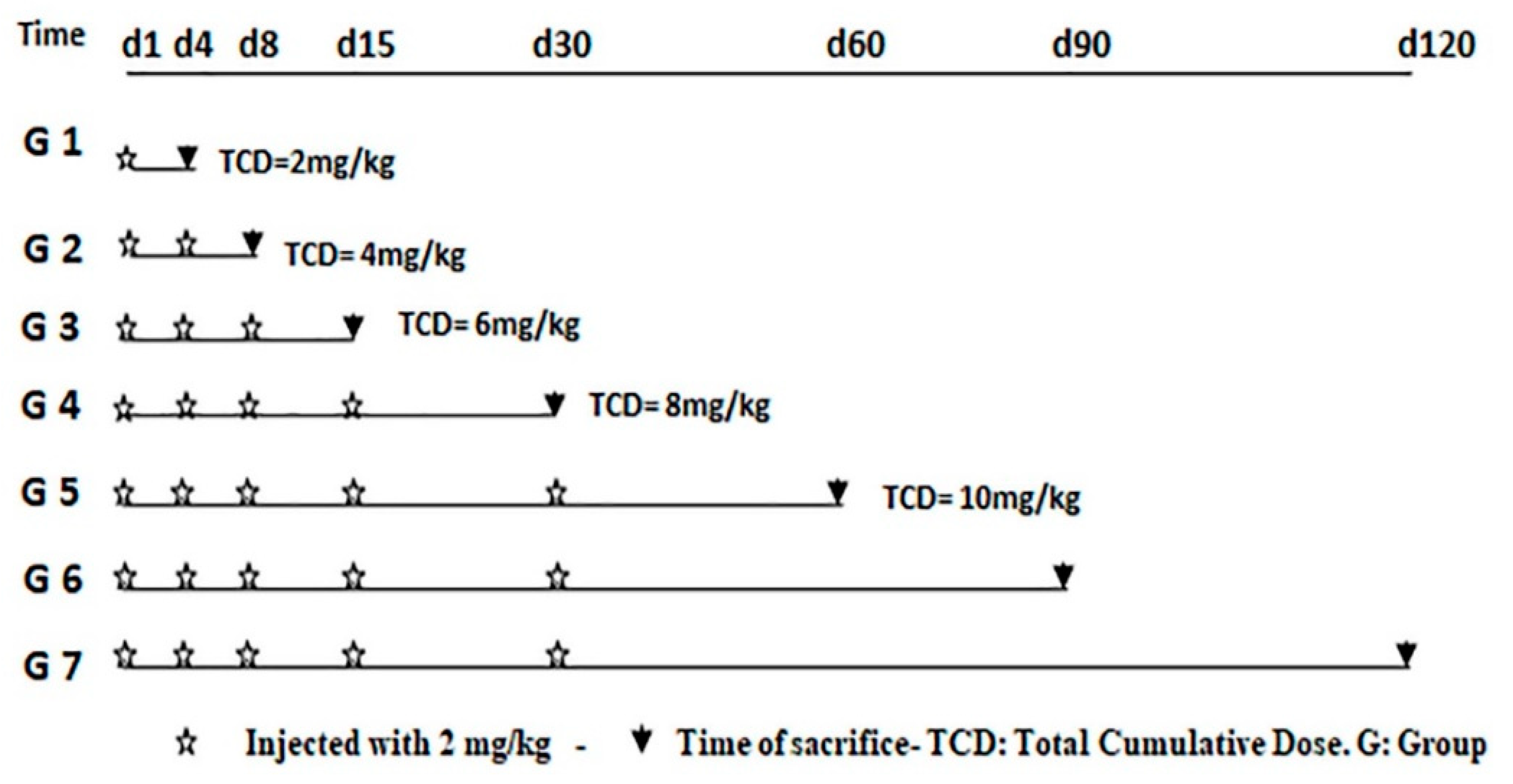
2.2.1. Experimental Design and Schedule of Sacrifice
2.2.2. Blood/Serum Collection and Analysis
2.2.3. Serum Biochemical and Antioxidant Analysis
2.2.4. Histopathological and Ultrastructural Examination
Histopathological Examination
Ultrastructural Examination
2.2.5. Statistical Analysis:
3. Results
3.1. Doxorubicin Induced Sgnificant Changes in Biochemical and Antioxidant Enzymes
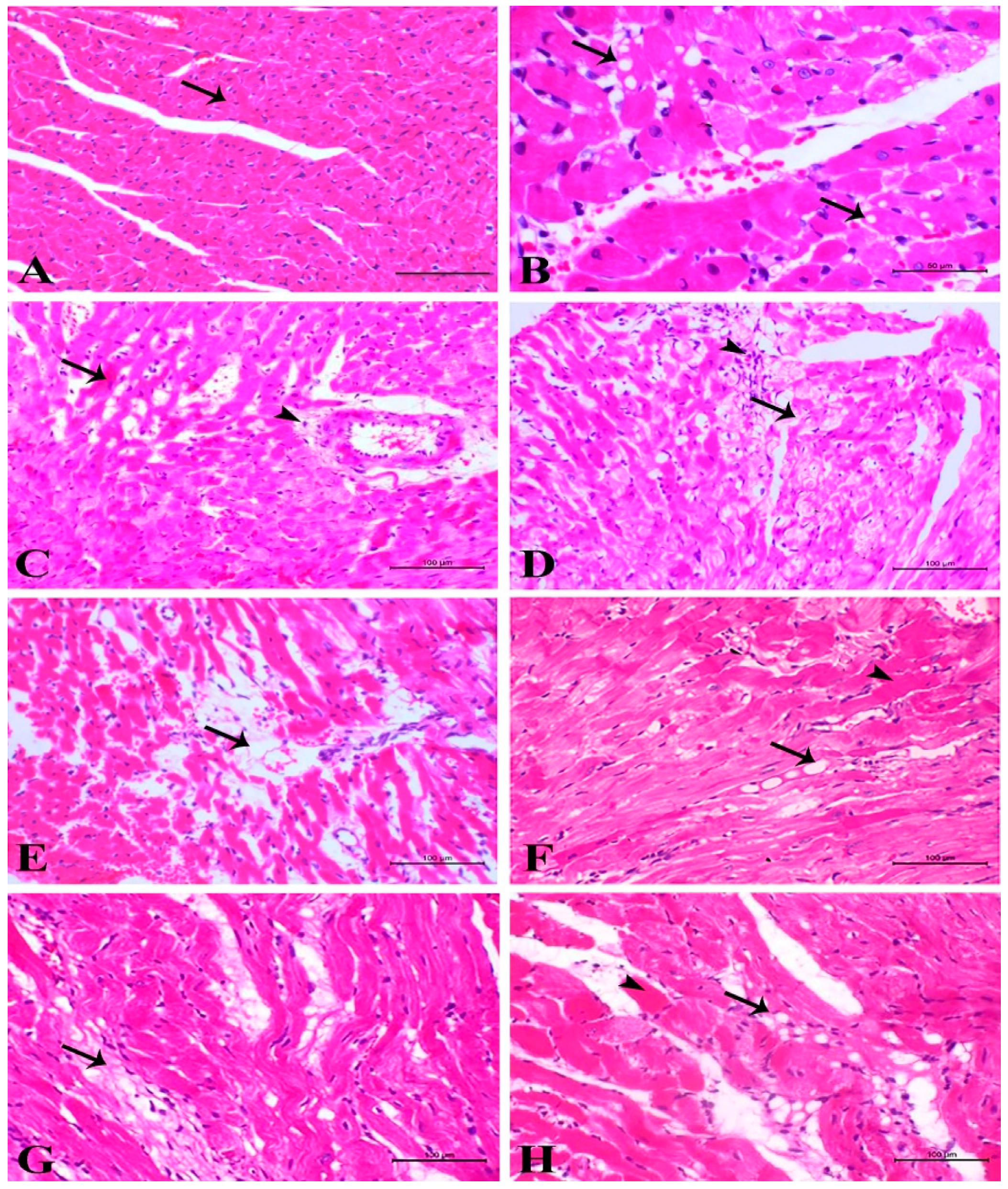
3.2. Light Microscopic Examinations of Cardiac Myocytes
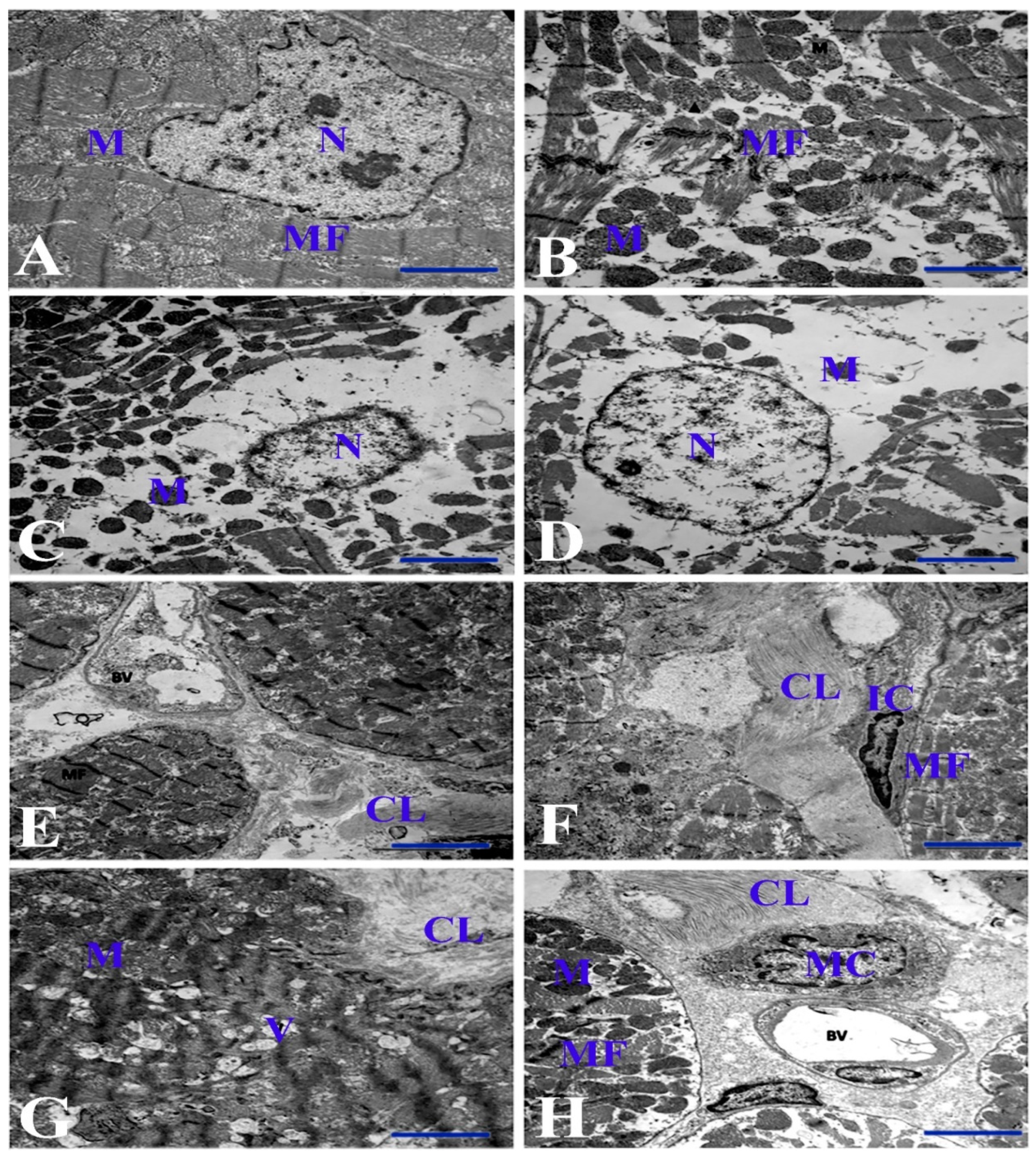
3.3. Electron Microscopic Examinations of Cardiac Myocytes and Their Mitochondria
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Booser, D.J.; Hortobagyi, G.N. Anthracycline Antibiotics in Cancer Therapy. Drugs 1994, 47, 223–258. [Google Scholar] [CrossRef]
- Johnson-Arbor, K.; Patel, H.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Al-Malky, H.S.; Osman, A.M.; Damanhouri, Z.A.; Alkreathy, H.M.; Al Aama, J.Y.; Ramadan, W.S.; Al Qahtani, A.A.; Al Mahdi, H.B. Modulation of doxorubicin-induced expression of the multidrug resistance gene in breast cancer cells by diltiazem and protection against cardiotoxicity in experimental animals. Cancer Cell Int. 2019, 19, 191. [Google Scholar] [CrossRef]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014, 1845, 84–89. [Google Scholar] [CrossRef]
- Fernandez-Chas, M.; Curtis, M.J.; A Niederer, S. Mechanism of doxorubicin cardiotoxicity evaluated by integrating multiple molecular effects into a biophysical model. Br. J. Pharmacol. 2018, 175, 763–781. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Lipsitz, S.R.; Sallan, S.E.; Dalton, V.M.; Mone, S.M.; Gelber, R.D.; Colan, S.D. Chronic Progressive Cardiac Dysfunction Years After Doxorubicin Therapy for Childhood Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2005, 23, 2629–2636. [Google Scholar] [CrossRef] [PubMed]
- Iarussi, D.; Indolfi, P.; Casale, F.; Martino, V.; Di Tullio, M.T.; Calabrò, R. Anthracycline-Induced Cardiotoxicity in Children with Cancer. Pediatr. Drugs 2005, 7, 67–76. [Google Scholar] [CrossRef]
- Aleman, B.M.P.; Belt-Dusebout, A.W.V.D.; De Bruin, M.L.; Veer, M.B.V.; Baaijens, M.H.A.; de Boer, J.P.; Hart, A.A.M.; Klokman, W.J.; Kuenen, M.A.; Ouwens, G.M.; et al. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood 2006, 109, 1878–1886. [Google Scholar] [CrossRef]
- Fonseca, A.; Coelho, P. Update on Biomarkers Associated to Cardioembolic Stroke: A Narrative Review. Life 2021, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Kehl, D.W.; Iqbal, N.; Fard, A.; Kipper, B.A.; Landa, A.D.L.P.; Maisel, A.S. Biomarkers in acute myocardial injury. Transl. Res. 2012, 159, 252–264. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffré, S.; Achilli, F.; Colombo, G.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Hear. Fail. Rev. 2017, 23, 109–122. [Google Scholar] [CrossRef]
- Herrmann, J. Clinical Cardio-Oncology, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2016; p. xxi. 491p. [Google Scholar]
- Dorr, R.T.; Lagel, K.; McLean, S. Cardioprotection of rat heart myocytes with amifostine (Ethyol®) and its free thiol, WR-1065, in vitro. Eur. J. Cancer 1996, 32, S21–S25. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Shi, J.; Li, Y.-J.; Wei, L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch. Immunol. Ther. Exp. 2009, 57, 435–445. [Google Scholar] [CrossRef]
- Liu, X.; Chua, C.C.; Gao, J.; Chen, Z.; Landy, C.L.C.; Hamdy, R.; Chua, B.H.L. Pifithrin-α protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am. J. Physiol. Circ. Physiol. 2004, 286, H933–H939. [Google Scholar] [CrossRef] [PubMed]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef]
- Wang, J.; Reijmers, T.; Chen, L.; Van Der Heijden, R.; Wang, M.; Peng, S.; Hankemeier, T.; Xu, G.; Van Der Greef, J. Systems toxicology study of doxorubicin on rats using ultra performance liquid chromatography coupled with mass spectrometry based metabolomics. Metabolomics 2009, 5, 407–418. [Google Scholar] [CrossRef]
- Zhao, L.-Q.; Zhang, B.-L. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef]
- Cove-Smith, L.; Woodhouse, N.; Hargreaves, A.; Kirk, J.; Smith, S.; Price, S.A.; Galvin, M.; Betts, C.J.; Brocklehurst, S.; Back-en, A.; et al. An Integrated Characterization of Serological, Pathological, and Functional Events in Doxorubicin-Induced Cardiotoxicity. Toxicol. Sci. 2014, 140, 3–15. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2019, 29, 101394. [Google Scholar] [CrossRef]
- Shaker, R.A.; Abboud, S.H.; Assad, H.C.; Hadi, N. Enoxaparin attenuates doxorubicin induced cardiotoxicity in rats via interfering with oxidative stress, inflammation and apoptosis. BMC Pharmacol. Toxicol. 2018, 19, 3. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ganame, J.; Claus, P.; Eyskens, B.; Uyttebroeck, A.; Renard, M.; D’Hooge, J.; Gewillig, M.; Bijnens, B.; Sutherland, G.R.; Mertens, L. Acute Cardiac Functional and Morphological Changes After Anthracycline Infusions in Children. Am. J. Cardiol. 2007, 99, 974–977. [Google Scholar] [CrossRef]
- Al-Saleem, I.A.; Jumaa, H.J.; Al-Ani, I.M.; Ismael, H.K. Morphological Changes in the Liver of Rats (Rattus norvegicus) treated with different Doses of Doxorubicin. Ann. Microsc. 2017, 16, 29–36. [Google Scholar]
- Herman, E.H.; Rahman, A.; Ferrans, V.J.; A Vick, J.; Schein, P.S. Prevention of chronic doxorubicin cardiotoxicity in beagles by liposomal encapsulation. Cancer Res. 1983, 43, 5427–5432. [Google Scholar]
- Alderton, P.M.; Gross, J.; Green, M.D. Comparative study of doxorubicin, mitoxantrone, and epirubicin in combination with ICRF-187 (ADR-529) in a chronic cardiotoxicity animal model. Cancer Res. 1992, 52, 194–201. [Google Scholar] [PubMed]
- Hayward, R.; Hydock, D.S. Doxorubicin cardiotoxicity in the rat: An in vivo characterization. J. Am. Assoc. Lab. Anim. Sci. 2007, 46, 20–32. [Google Scholar]
- Olson, H.M.; Capen, C.C. Chronic cardiotoxicity of doxorubicin (Adriamycin) in the rat: Morphologic and biochemical investigations. Toxicol. Appl. Pharmacol. 1978, 44, 605–616. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Volkova, M. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 2012, 7, 214–220. [Google Scholar] [CrossRef]
- Umlauf, J.; Horký, M. Molecular biology of doxorubicin-induced cardiomyopathy. Exp. Clin. Cardiol. 2002, 7, 35–39. [Google Scholar]
- Doroshow, J.H.; Locker, G.Y.; Myers, C.E. Enzymatic Defenses of the Mouse Heart against Reactive Oxygen Metabolites. J. Clin. Investig. 1980, 65, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Sazuka, Y.; Tanizawa, H.; Takino, Y. Effect of Adriamycin on the Activities of Superoxide Dismutase, Glutathione Peroxidase and Catalase in Tissues of Mice. Jpn. J. Cancer Res. 1989, 80, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Danelisen, I.; Singal, P.K. Early changes in myocardial antioxidant enzymes in rats treated with adriamycin. Mol. Cell. Biochem. 2002, 232, 19–26. [Google Scholar] [CrossRef]
- Lebrecht, D.; Kokkori, A.; Setzer, B.; Walker, U.A.; Ketelsen, U.-P. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J. Pathol. 2005, 207, 436–444. [Google Scholar] [CrossRef]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef]
- Santos, D.L.; Moreno, A.J.; Leino, R.L.; Froberg, M.K.; Wallace, K.B. Carvedilol Protects against Doxorubicin-Induced Mitochondrial Cardiomyopathy. Toxicol. Appl. Pharmacol. 2002, 185, 218–227. [Google Scholar] [CrossRef]
- Abeer, E.E.; Mansour, K.F.; Nariman, A.A.; Fatma, A.E.; El-Fiky, M.M. Histological and immunohistochemical study on the effect of doxorubicin on the heart of adult albino rat and the possible protective role of an antioxidant. Menoufiya Med. J. 2008, 21, 91–108. [Google Scholar]
- Lüpertz, R.; Wätjen, W.; Kahl, R.; Chovolou, Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 2010, 271, 115–121. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Sahota, P.S.; Popp, J.A.; Hardisty, J.F.; Gopinath, C.; Bouchard, P. Toxicologic Pathology: Nonclinical Safety Assessment, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2018; p. xix. 1224p. [Google Scholar]
- Sonneveld, P. Pharmacokinetics of Adriamycin in the Rat. Ph.D. Thesis, Delft University of Technology, Leiden, The Netherlands, 1980; p. 125. [Google Scholar]
- Villani, F.; Galimberti, M.; Zunino, F.; Monti, E.; Rozza, A.; Lanza, E.; Favalli, L.; Poggi, P. Prevention of doxorubicin-induced cardiomyopathy by reduced glutathione. Cancer Chemother. Pharmacol. 1991, 28, 365–369. [Google Scholar] [CrossRef]
- Xu, M.; Sheng, L.-H.; Zhu, X.-H.; Zeng, S.-B.; Chi, D.-X.; Zhang, G.-J. Protective Effect of Tetrandrine on Doxorubicin-Induced Cardiotoxicity in Rats. Tumori 2010, 96, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Balazs, T.; Ferrans, V.J. Cardiac Lesions Induced by Chemicals. Environ. Heal. Perspect. 1978, 26, 181. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Vivar, J.; Martasek, P.; Hogg, N.; Masters, B.S.S.; Pritchard, K.A.; Kalyanaraman, B. Endothelial Nitric Oxide Synthase-Dependent Superoxide Generation from Adriamycin. Biochemistry 1997, 36, 11293–11297. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Kotamraju, S.; Zhao, H.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced Apoptosis Is Associated with Increased Transcription of Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2001, 276, 47266–47276. [Google Scholar] [CrossRef] [PubMed]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of Nitric Oxide Synthase 3 Protects Against the Cardiac Injury, Dysfunction, and Mortality Induced by Doxorubicin. Circulation 2007, 116, 506–514. [Google Scholar] [CrossRef]
- Xu, M.F.; Tang, P.L.; Qian, Z.M.; Ashraf, M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci. 2001, 68, 889–901. [Google Scholar] [CrossRef]
- Minotti, G.; Ronchi, R.; Salvatorelli, E.; Menna, P.; Cairo, G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: Evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Res. 2001, 61, 8422–8428. [Google Scholar]
- Šimůnek, T.; Štěrba, M.; Popelová, O.; Adamcova, M.; Hrdina, R.; Geršl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Prasad, S.V.N.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Bertinchant, J.P.; Polge, A.; Juan, J.M.; Oliva-Lauraire, M.C.; Giuliani, I.; Marty-Double, C.; Burdy, J.Y.; Fabbro-Peray, P.; Laprade, M.; Bali, J.P.; et al. Evaluation of cardiac troponin I and T levels as markers of myocardial damage in doxorubicin-induced cardiomyopathy rats, and their relationship with echocardiographic and histological findings. Clin. Chim. Acta. 2003, 329, 39–51. [Google Scholar] [CrossRef]
- Balli, E.; O Mete, U.; Tuli, A.; Tap, O.; Kaya, M. Effect of melatonin on the cardiotoxicity of doxorubicin. Histol. Histopathol. 2004, 19, 1101–1108. [Google Scholar] [CrossRef]
- Yilmaz, S.; Ateşşahin, A.; Sahna, E.; Karahan, I.; Ozer, S. Protective effect of lycopene on adriamycin-induced cardiotoxicity and nephrotoxicity. Toxicology 2006, 218, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M.; Casini, A.; Albano, E.; Poli, G.; Gentilini, A.; Dianzani, M. Stimulation of Lipid Peroxidation or 4-Hydroxynonenal Treatment Increases Procollagen α1 (I) Gene Expression in Human Liver Fat-Storing Cells. Biochem. Biophys. Res. Commun. 1993, 194, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Parola, M. Oxidative damage and fibrogenesis. Free Radic. Biol. Med. 1997, 22, 287–305. [Google Scholar] [CrossRef]
- Fan, J.; Zeng, M.; Li, J. Correlation between hepatic fat, lipid peroxidation and hepatic fibrosis in rats chronically fed with ethanol and/or high fat diet. Zhonghua Nei Ke Za Zhi 1997, 36, 808–811. [Google Scholar] [PubMed]
- Ore, A.; Akinloye, O. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Dubey, K.; Anwer, T.; Ashish, A.; Pillai, K.K. Protective effects of telmisartan against acute doxorubicin-induced cardiotoxicity in rats. Pharmacol. Rep. 2008, 60, 382. [Google Scholar] [PubMed]
- Saraogi, P.; Pillai, K.K.; Singh, B.K.; Dubey, K. RETRACTED: Rosiglitazone and pioglitazone aggravate doxorubicin-induced cardiomyopathy in Wistar rats. Biomed. Pharmacother 2010, 1, 65–71. [Google Scholar] [CrossRef]
- Olson, H.M.; Young, D.M.; Prieur, D.J.; Leroy, A.F.; Reagan, R.L. Electrolyte and Morphologic Alterations of Myocardium in Adriamycin-Treated Rabbits. Am. J. Pathol. 1974, 77, 439–454. [Google Scholar]
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J. Evidence of a complex between adriamycin derivatives and cardiolipin: Possible role in cardiotoxicity. Biochem. Pharmacol. 1980, 29, 3003–3010. [Google Scholar] [CrossRef]
- Fadillioglu, E.; Oztas, E.; Erdogan, H.; Yagmurca, M.; Sogut, S.; Ucar, M.; Irmak, M.K. Protective effects of caffeic acid phenethyl ester on doxorubicin-induced cardiotoxicity in rats. J. Appl. Toxicol. 2004, 24, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Hasinoff, B.B. Inhibition and inactivation of NADH-cytochrome c reductase activity of bovine heart submitochondrial particles by the iron(III)-adriamycin complex. Biochem. J. 1990, 265, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, K.; Molgaard, K.; Steiness, E. Similar changes in cardiac morphology and DNA synthesis induced by doxorubicin and 4′-epi-doxorubicin. Cancer Chemother. Pharmacol. 1985, 15, 244–252. [Google Scholar] [CrossRef]
- Palmeira, C.; Serrano, J.; Kuehl, D.; Wallace, K. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim. Biophys. Acta (BBA) Bioenerg. 1997, 1321, 101–106. [Google Scholar] [CrossRef]
- L’Ecuyer, T.; Sanjeev, S.; Thomas, R.; Novak, R.; Das, L.; Campbell, W.; Heide, R.V. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am. J. Physiol. Circ. Physiol. 2006, 291, H1273–H1280. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Phungphong, S.; Kijtawornrat, A.; Kampaengsri, T.; Wattanapermpool, J.; Bupha-Intr, T. Comparison of exercise training and estrogen supplementation on mast cell-mediated doxorubicin-induced cardiotoxicity. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R829–R842. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Groups | AST (U/L) | CK–MB (U/L) | cTnT (µg/L) | Catalase (U/mg Protein) | SOD (mU/mg Protein) | GPx (mU/mg Protein) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control (n = 3) | Treated (n = 5) | Control (n = 3) | Treated (n = 5) | Control (n = 3) | Treated (n = 5) | Control (n = 3) | Treated (n = 5) | Control (n = 3) | Treated (n = 5) | Control (n = 3) | Treated (n = 5) | |
| G1 | 57.50 ± 3.12 | 74.80 ± 5.89 a | 304.40 ± 14.38 | 410.40 ± 9.40 a | 10.64 ± 2.12 | 16.85 ± 3.04 a | 57.32 ± 4.75 | 44.97 ± 4.56 a | 41.45 ± 2.34 | 31.96 ± 3.47 a | 29.11 ± 1.39 | 19.47 ± 2.64 a |
| G2 | 55.30 ± 3.13 | 89.60 ± 6.39 a | 306.40 ± 22.33 | 532.40 ± 14.67 a | 10.70 ± 2.27 | 28.09 ± 2.18 a | 56.13 ± 4.75 | 36.08 ± 3.63 a | 40.37 ± 3.23 | 25.60 ± 3.62 a | 28.20 ± 1.30 | 19.21 ± 3.19 a |
| G3 | 57.40 ± 2.43 | 130.00 ± 8.28 a,b,c | 309.38 ± 16.27 | 809.80 ± 32.87 a,b,c | 10.92 ± 1.75 | 40.34 ± 3.36 a,b,c | 53.22 ± 4.75 | 26.67 ± 2.83 a,b,c | 39.63 ± 2.20 | 22.02 ± 2.15 a,b,c | 28.62 ± 2.12 | 16.68 ± 2.58 a,b |
| G4 | 58.50 ± 3.18 | 157.00 ± 5.66 a,b,c,d | 310.40 ± 23.11 | 900.20 ± 14.06 a,b,c,d | 11.36 ± 2.47 | 52.96 ± 3.66 a,b,c,d | 54.40 ± 4.75 | 20.05 ± 1.52 a,b,c | 40.34 ± 2.69 | 20.88 ± 4.38 a,b,c | 27.25 ± 1.04 | 12.93 ± 1.89 a,b,c |
| G5 | 60.20 ± 2.27 | 117.20 ± 4.97 a,b,c,e | 311.38 ± 25.24 | 688.00 ± 19.95 a,b,c,e | 11.46 ± 2.23 | 38.54 ± 1.49 a,b,c,e | 56.62 ± 4.75 | 20.94 ± 3.45 a,b,c | 39.23 ± 2.34 | 15.12 ± 3.29 a,b,c,e | 28.75 ± 2.50 | 12.07 ± 2.88 a,b,c |
| G6 | 63.80 ± 3.36 | 104.60 ± 7.44 a,b,c,d | 315.40 ± 21.74 | 610.80 ± 23.95 a,b,d | 12.13 ± 2.44 | 28.42 ± 2.42 a,b,d,e,f | 52.25 ± 4.75 | 32.76 ± 3.95 a,b,e,f | 38.14 ± 1.18 | 22.22 ± 2.96 a,b | 26.90 ± 1.18 | 14.54 ± 0.97 a,b |
| G7 | 62.30 ± 3.38 | 89.80 ± 10.76 a,f,g | 321.38 ± 18.19 | 589.00 ± 11.58 a,f | 13.83 ± 2.26 | 22.38 ± 3.11 a,f | 50.13 ± 4.75 | 39.06 ± 2.37 a,f | 38.25 ± 3.16 | 30.50 ± 3.83 a,f | 25.85 ± 2.38 | 17.05 ± 2.05 a,f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelatty, A.; Ahmed, M.S.; Abdel-Kareem, M.A.; Dmerdash, M.; Mady, R.; Saad, A.S.; Albrakati, A.; Elmahallawy, E.K.; Elsawak, A.; Abdo, W. Acute and Delayed Doxorubicin-Induced Myocardiotoxicity Associated with Elevation of Cardiac Biomarkers, Depletion of Cellular Antioxidant Enzymes, and Several Histopathological and Ultrastructural Changes. Life 2021, 11, 880. https://doi.org/10.3390/life11090880
Abdelatty A, Ahmed MS, Abdel-Kareem MA, Dmerdash M, Mady R, Saad AS, Albrakati A, Elmahallawy EK, Elsawak A, Abdo W. Acute and Delayed Doxorubicin-Induced Myocardiotoxicity Associated with Elevation of Cardiac Biomarkers, Depletion of Cellular Antioxidant Enzymes, and Several Histopathological and Ultrastructural Changes. Life. 2021; 11(9):880. https://doi.org/10.3390/life11090880
Chicago/Turabian StyleAbdelatty, Alaa, Mohamed S. Ahmed, Mona A. Abdel-Kareem, Mohamed Dmerdash, Rehab Mady, Ahmed S. Saad, Ashraf Albrakati, Ehab Kotb Elmahallawy, Ahmed Elsawak, and Walied Abdo. 2021. "Acute and Delayed Doxorubicin-Induced Myocardiotoxicity Associated with Elevation of Cardiac Biomarkers, Depletion of Cellular Antioxidant Enzymes, and Several Histopathological and Ultrastructural Changes" Life 11, no. 9: 880. https://doi.org/10.3390/life11090880
APA StyleAbdelatty, A., Ahmed, M. S., Abdel-Kareem, M. A., Dmerdash, M., Mady, R., Saad, A. S., Albrakati, A., Elmahallawy, E. K., Elsawak, A., & Abdo, W. (2021). Acute and Delayed Doxorubicin-Induced Myocardiotoxicity Associated with Elevation of Cardiac Biomarkers, Depletion of Cellular Antioxidant Enzymes, and Several Histopathological and Ultrastructural Changes. Life, 11(9), 880. https://doi.org/10.3390/life11090880