
Identification of Targeted Proteins by Jamu Formulas for Different Efficacies Using Machine Learning Approach

 , ,
, , 


Abstract
:1. Introduction
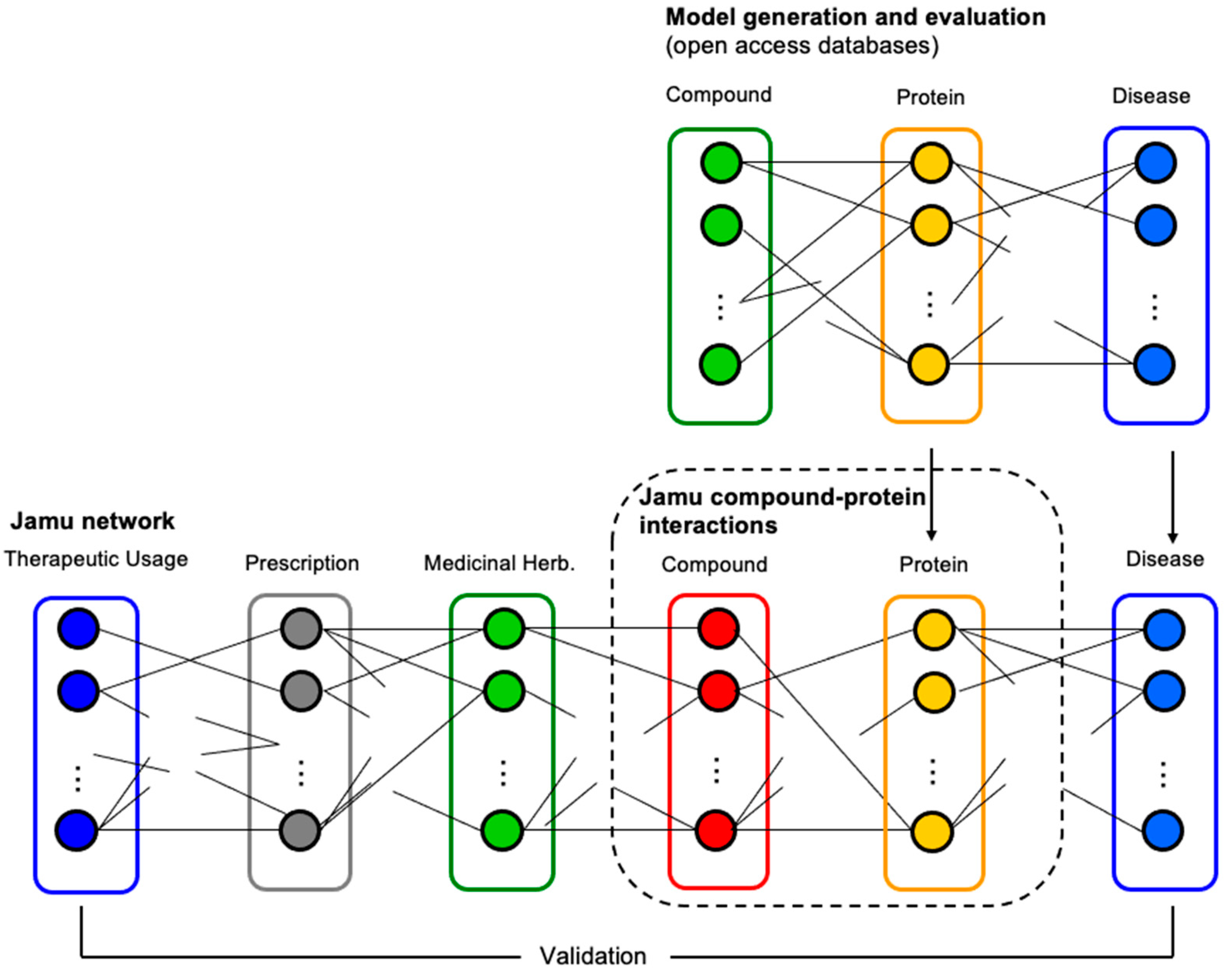
2. Materials and Methods
2.1. Data Transformation
2.2. Model Generation and Evaluation
2.3. Prediction of the Target Protein by Jamu Formulas
3. Results and Discussion
3.1. Preprocessing of Compound and Protein Data
3.2. Prediction Performance
3.3. Prediction Results
4. Conclusions and Future Works
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Sheng, Y.; Wang, Q.; Amin, A.; Li, X.; Xie, Y. Potential compound from herbal food of Rhizoma Polygonati for treatment of COVID-19 analyzed by network pharmacology: Viral and cancer signaling mechanisms. J. Funct. Foods 2021, 77, 104149. [Google Scholar] [CrossRef]
- Chen, W.; Lim, C.E.D.; Kang, H.-J.; Liu, J. Chinese herbal medicines for the treatment of type A H1N1 influenza: A systematic review of randomized controlled trials. PLoS ONE 2011, 6, e028093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safarzadeh, E.; Shotorbani, S.S.; Baradaran, B. Herbal medicine as inducers of apoptosis in cancer treatment. Adv. Pharm. Bull. 2014, 4, 421–427. [Google Scholar] [CrossRef]
- Schippmann, U.; Leaman, D.J.; Cunningham, A.B. Impact of cultivation and gathering of medicinal plants on biodiversity: Global trends and issues. Biodivers. Ecosyst. Approach Agric. For. Fish. 2002, 1–21. [Google Scholar] [CrossRef]
- Schippmann, U.; Leaman, D.; Cunningham, A. A comparison of cultivation and wild collection of medicinal and aromatic plants under sustainability aspects. In Medicinal and Aromatic Plants; Springer: Dordrecht, The Netherlands, 2006; pp. 75–95. ISBN 9783540563914. [Google Scholar]
- Hanafi, M.; Nina, A.; Fadia, Z.; Nurbaiti, N. Indonesian Country Report on Traditional Medicine; CSIR: New Delhi, India, 2006. [Google Scholar]
- Furnham, A. Why do people choose and use complementary therapies. In Complementary Medicine: An Objective Appraisal; Ernst, E., Ed.; Butterworth-Heinemann: Oxford, UK, 1996; pp. 71–88. [Google Scholar]
- Ernst, E. Herbal medicines put into context: Their use entails risks, but probably fewer than with synthetic drugs. BMJ Br. Med. J. 2003, 327, 881. [Google Scholar] [CrossRef]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef]
- Wijaya, S.H.; Batubara, I.; Nishioka, T.; Altaf-Ul-Amin, M.; Kanaya, S. Metabolomic studies of Indonesian Jamu medicines: Prediction of Jamu efficacy and identification of important metabolites. Mol. Inform. 2017, 36, 1700050. [Google Scholar] [CrossRef] [PubMed]
- Nasution, A.K.; Wijaya, S.H.; Kusuma, W.A. Prediction of drug-target interaction on Jamu formulas using machine learning approaches. In Proceedings of the ICACSIS 2019: 11th International Conference on Advanced Computer Science and Information Systems, Nusa Dua, Indonesia, 12–13 October 2019; pp. 169–174. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; MacIejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, 1091–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, 514–517. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, 232–240. [Google Scholar] [CrossRef]
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Bleakley, K.; Yamanishi, Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 2009, 25, 2397–2403. [Google Scholar] [CrossRef] [Green Version]
- Gunn, S.R. Support Vector Machines for Classification and Regression; University of Southampton: Southampton, UK, 1998; Volume 14. [Google Scholar]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. In Annual Reports in Computational Chemistry; Wheeler, R.A., Spellmeyer, D.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 217–241. [Google Scholar]
- Gunther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36, 919–922. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank and its relevance to pharmacogenomics. Pharmacogenomics 2008, 9, 1155–1162. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. Protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [Green Version]
- Vapnik, V. Statistical Learning Theory (Adaptive and Cognitive Dynamic Systems: Signal Processing, Learning, Communications and Control); John Wiley & Sons: Hoboken, NJ, USA, 1998; pp. 1–740. [Google Scholar]
- Hussain, M.; Wajid, S.K.; Elzaart, A.; Berbar, M. A comparison of SVM kernel functions for breast cancer detection. In Proceedings of the 2011 8th International Conference on Computer Graphics, Imaging and Visualization (CGIV 2011), Singapore, 17–19 August 2011; pp. 145–150. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Tang, W.; Wu, X.; Fu, W. A random forest approach to the detection of epistatic interactions in case-control studies. BMC Bioinform. 2009, 10, S65. [Google Scholar] [CrossRef] [Green Version]
- Duda, R.O.; Hart, P.E.; Stork, D.G. Pattern Classification; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Deng, H.; Runger, G. Gene selection with guided regularized random forest. Pattern Recognit. 2013, 46, 3483–3489. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Zeng, N.; Wang, N. Sensitivity, specificity, accuracy, associated confidence interval and ROC analysis with practical SAS® implementations. In Proceedings of the NESUG: Health Care and Life Sciences, Baltimore, MA, USA, 14–17 November 2010; pp. 1–9. [Google Scholar]
- Wijaya, S.H.; Husnawati, H.; Afendi, F.M.; Batubara, I.; Darusman, L.K.; Altaf-Ul-Amin, M.; Sato, T.; Ono, N.; Sugiura, T.; Kanaya, S. Supervised clustering based on DPClusO: Prediction of plant-disease relations using Jamu formulas of KNApSAcK database. Biomed Res. Int. 2014, 2014, 831751. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A. e1071: Misc Functions of the Department of Statistics (e1071); R Package Version 1(3); TU Wien: Vienna, Austria, 2014; pp. 1–62. [Google Scholar]
- Yang, S.; Zhu, F.; Ling, X.; Liu, Q.; Zhao, P. Intelligent health care: Applications of deep learning in computational medicine. Front. Genet. 2021, 12, 607471. [Google Scholar] [CrossRef]
- Jurasekova, Z.; Marconi, G.; Sanchez-Cortes, S.; Torreggiani, A. Spectroscopic and molecular modeling studies on the binding of the flavonoid luteolin and human serum albumin. Biopolymers 2009, 91, 917–927. [Google Scholar] [CrossRef]
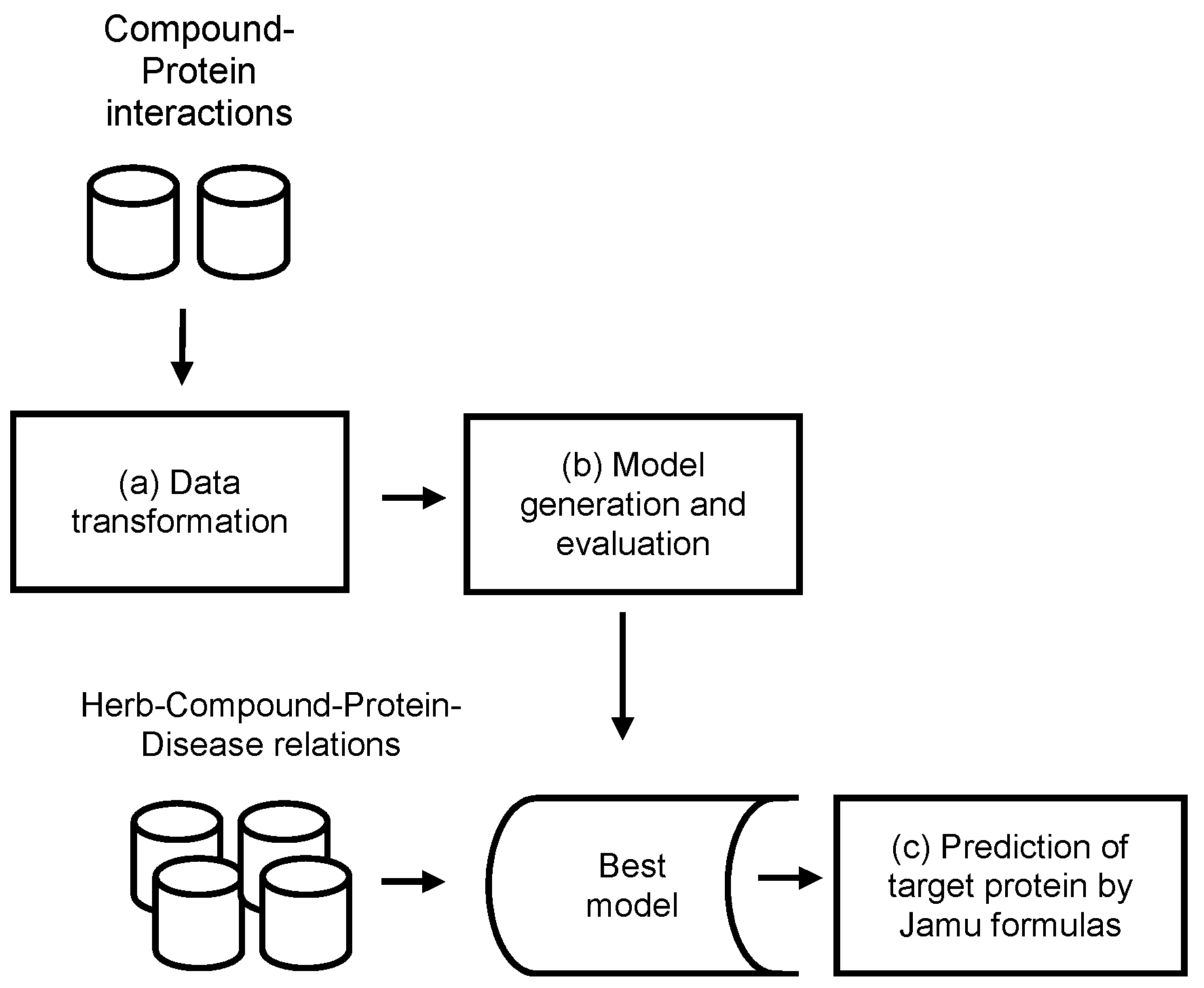


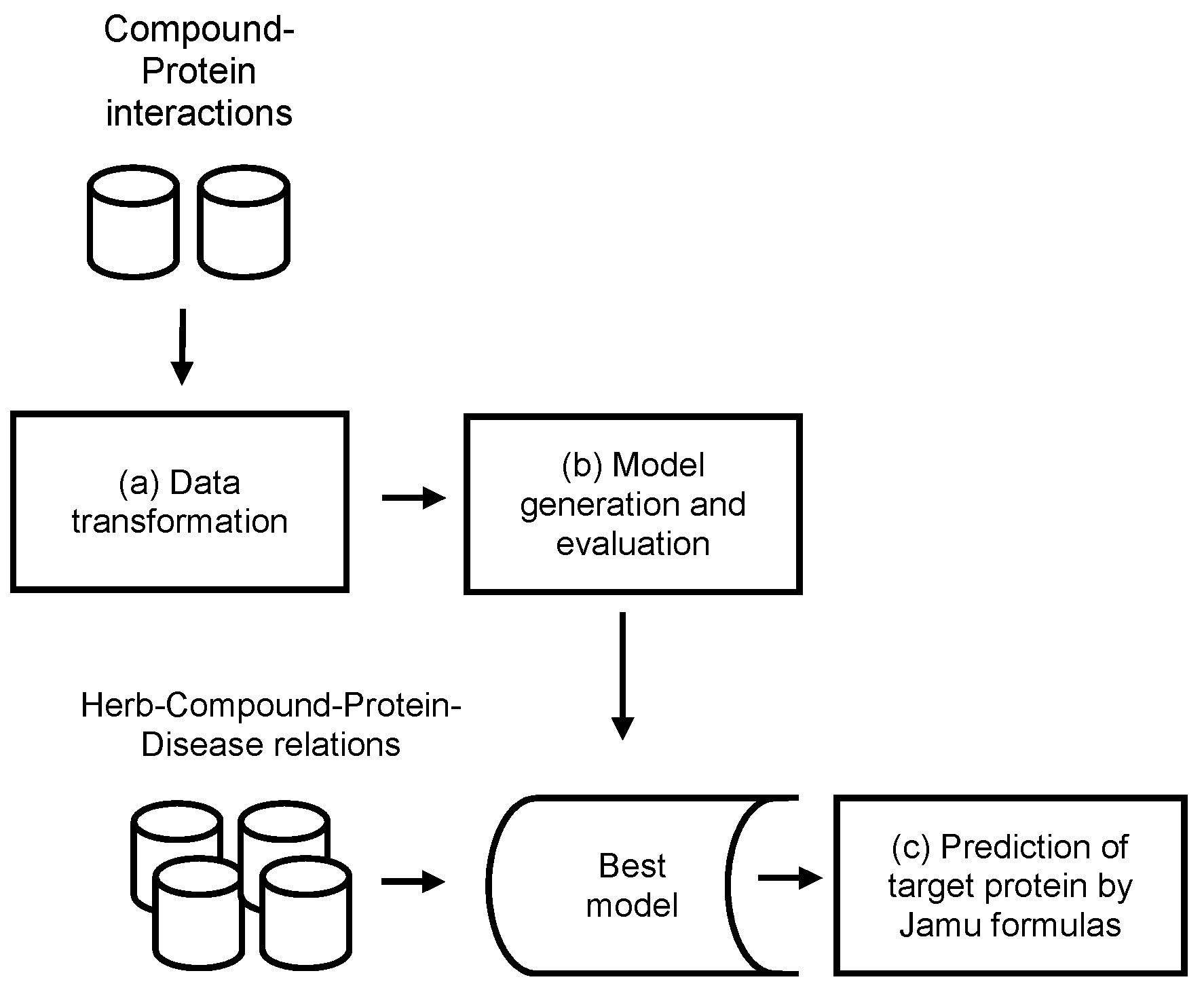{kind=link}
{kind=link}
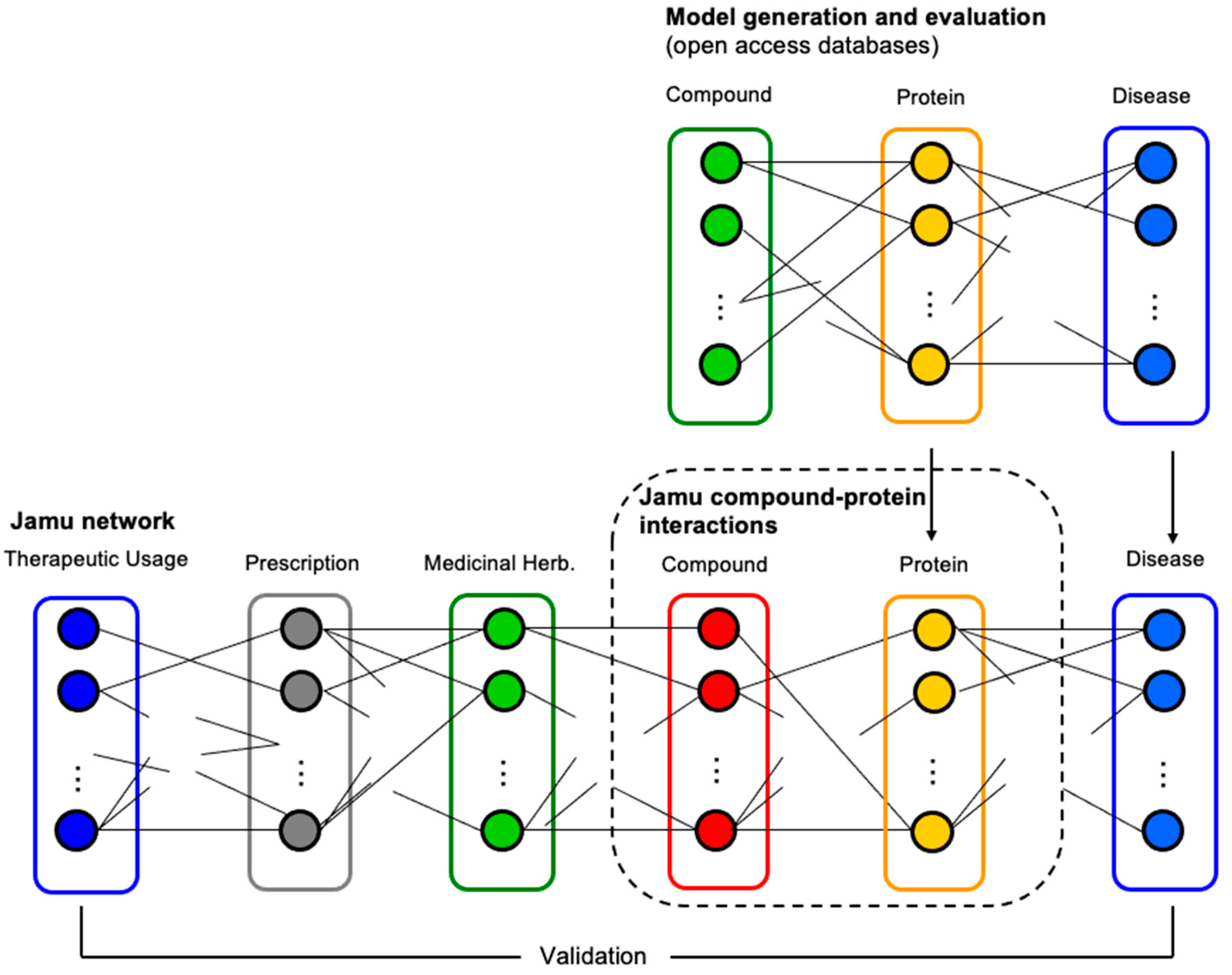{kind=link}
| Description | Number of Data | Identifier | References |
|---|---|---|---|
| Protein | 3.334 | UniProtID | UniProt, IJAH |
| Compound | 17.277 | CAS_ID, PubChem ID, KEGG ID | KNApSAcK, PubChem, KEGG, IJAH |
| Compound of Jamu | 94 | Compound ID | Wijaya et al. [11] |
| Compound–protein interactions | 149 | KEGG | |
| 4.144 | Matador | ||
| 3.696 | UniProt, IJAH | ||
| Amino acid sequences | 3.334 | UniProtID | UniProt |
| ID | Efficacy Groups | Number of Compounds |
|---|---|---|
| E1 | Blood and Lymph Diseases | 15 |
| E2 | Cancers | 5 |
| E3 | The Digestive System | 17 |
| E4 | Female-Specific Diseases | 16 |
| E5 | The Heart and Blood Vessels | 4 |
| E6 | Male-Specific Diseases | 5 |
| E7 | Muscle and Bone | 18 |
| E8 | Nutritional and Metabolic Diseases | 7 |
| E9 | Respiratory Diseases | 32 |
| E10 | Skin and Connective Tissue | 4 |
| E11 | The Urinary System | 14 |
| E12 | Mental and Behavioral Disorders | 8 |
| Datasets | Classifiers | Accuracy | Sensitivity | Specificity | |||
|---|---|---|---|---|---|---|---|
| MACCS_AAC | SVM Linear | 69.16%± | 1.07% | 71.52%± | 1.84% | 66.77%± | 1.87% |
| SVM RBF | 81.71%± | 1.52% | 82.79%± | 2.27% | 80.62%± | 1.27% | |
| Random Forest | 89.30%± | 0.69% | 87.86%± | 1.20% | 90.74%± | 1.05% | |
| MACCS_Dipeptide | SVM Linear | 61.68%± | 0.77% | 63.49%± | 1.61% | 61.27%± | 0.88% |
| SVM RBF | 72.71%± | 0.86% | 71.81%± | 1.81% | 73.15%± | 1.15% | |
| Random Forest | 60.79%± | 1.20% | 59.14%± | 1.56% | 61.17%± | 1.30% | |
| PubChem_AAC | SVM Linear | 70.77%± | 0.90% | 73.08%± | 1.86% | 68.49%± | 1.87% |
| SVM RBF | 80.01%± | 1.35% | 80.52%± | 1.80% | 79.51%± | 1.82% | |
| Random Forest | 89.28%± | 0.40% | 87.96%± | 0.88% | 90.63%± | 0.58% | |
| PubChem_Dipeptide | SVM Linear | 50.49%± | 1.08% | 54.15%± | 1.38% | 50.47%± | 1.01% |
| SVM RBF | 49.55%± | 1.28% | 54.83%± | 5.44% | 49.56%± | 1.19% | |
| Random Forest | 50.28%± | 0.72% | 50.12%± | 1.60% | 50.28%± | 0.71% | |
| No | Compound ID | Compound Name | Molecular Formula | UniProt ID | Targeted Protein | OMIM ID | Disease Description |
|---|---|---|---|---|---|---|---|
| E1 Blood and Lymph Diseases | |||||||
| 1 | N/A | (4Z)-1-(2,3,5-Trihydroxy-4-methylphenyl)dec-4-en-1-one | C17H24O4 | P02768 | Serum albumin | 615999; 616000 | Hyperthyroxinemia, familial dysalbuminemic; analbuminemia |
| 2 | 689043, C00000615 | Caffeic acid | C9H8O4 | ||||
| 3 | 5317587, | Diacetoxy-[6]-gingerdiol | C21H32O6 | ||||
| 4 | 370, C00002647 | Gallic acid | C7H6O5 | ||||
| 5 | 5280445, C00000674 | Luteolin | C15H10O6 | ||||
| 6 | 5280459, C00005373 | Quercitrin | C21H20O11 | ||||
| 7 | 5281702, C00013329 | Tricin | C17H14O7 | ||||
| 8 | 64945, C00003558 | Ursolic acid | C30H48O3 | Q92887 | Canalicular multispecific organic anion transporter 1 | 237500 | Dubin–Johnson syndrome |
| Q9NPD5 | Solute carrier organic anion transporter family member 1B3 | 237450 | Hyperbilirubinemia, rotor type | ||||
| Q9Y6L6 | Solute carrier organic anion transporter family member 1B1 | Hyperbilirubinemia, rotor type | |||||
| P08185 | Corticosteroid-binding globulin | 611489 | Corticosteroid-binding globulin deficiency | ||||
| P02768 | Serum albumin | 615999; 616000 | Hyperthyroxinemia, familial dysalbuminemic; analbuminemia | ||||
| 9 | 73145, C00003738 | beta-Amyrin | C30H50O | Q92887 | Canalicular multispecific organic anion transporter 1 | 237500 | Dubin–Johnson syndrome |
| 10 | 222284, C00003672 | beta-Sitosterol | C29H50O | Dubin–Johnson syndrome | |||
| E3 The Digestive System | |||||||
| 1 | 519857, C00020146 | 1-epi-Cubenol | C15H26O | P08183 | Multidrug resistance protein 1 | 612244 | Inflammatory bowel disease 13 |
| 2 | N/A | Anisucumarin A | C20H20O8 | ||||
| 3 | 240, C00034452 | Benzaldehyde | C7H6O | ||||
| 4 | 6448, C00029844 | Bornyl acetate | C12H20O2 | ||||
| 5 | 3314, C00000619 | Eugenol | C10H12O2 | ||||
| 6 | 289151, C00003162 | Longifolene | C15H24 | ||||
| 7 | N/A | Morin-3-O-lyxoside | C20H18O11 | ||||
| 8 | 985, C00001233 | Palmitic acid | C16H32O2 | ||||
| 9 | 442402, C00003194 | Thujopsene | C15H24 | ||||
| 10 | 12306047, C00029671 | alpha-Muurolene | C15H24 | ||||
| 11 | 7460, C00003051 | alpha-Phellandrene | C10H16 | ||||
| 12 | 111037, C00035043 | alpha-Terpinyl acetate | C12H20O2 | ||||
| 13 | 12313020, C00020130 | gamma-Muurolene | C15H24 | ||||
| E4 Female-Specific Diseases | |||||||
| 1 | 5280794, C00003674 | Stigmasterol | C29H48O | P11511 | Aromatase | 139300; 613546 | Aromatase excess syndrome; aromatase deficiency |
| P03372 | Estrogen receptor | 615363 | Estrogen resistance | ||||
| E7 Muscle and Bone | |||||||
| 1 | 10131321, C00055009 | Coumaperine | C16H19NO2 | P20309 | Muscarinic acetylcholine receptor M3 | 100100 | Prune belly syndrome |
| E8 Nutritional and Metabolic Diseases | |||||||
| 1 | 3084331, C00020154 | T-Muurolol | C15H26O | Q92887 | Canalicular multispecific organic anion transporter 1 | 237500 | Dubin–Johnson syndrome |
| Q02318 | Sterol 26-hydroxylase, mitochondrial | 213700 | Cerebrotendinous xanthomatosis | ||||
| P11473 | Vitamin D3 receptor | 277440 | Rickets vitamin D-dependent 2A | ||||
| E10 Skin and Connective Tissue | |||||||
| 1 | 222284, C00003672 | beta-Sitosterol | C29H50O | Q02318 | Sterol 26-hydroxylase, mitochondrial | 213700 | Cerebrotendinous xanthomatosis |
| E12 Mental and Behavioral Disorders | |||||||
| 1 | 6989, C00000155 | Thymol | C10H14O | P08172 | Muscarinic acetylcholine receptor M2 | 608516 | Major depressive disorder |
| Q13002 | Glutamate receptor ionotropic, kainate 2 | 611092 | Mental retardation, autosomal recessive 6 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijaya, S.H.; Afendi, F.M.; Batubara, I.; Huang, M.; Ono, N.; Kanaya, S.; Altaf-Ul-Amin, M. Identification of Targeted Proteins by Jamu Formulas for Different Efficacies Using Machine Learning Approach. Life 2021, 11, 866. https://doi.org/10.3390/life11080866
Wijaya SH, Afendi FM, Batubara I, Huang M, Ono N, Kanaya S, Altaf-Ul-Amin M. Identification of Targeted Proteins by Jamu Formulas for Different Efficacies Using Machine Learning Approach. Life. 2021; 11(8):866. https://doi.org/10.3390/life11080866
Chicago/Turabian StyleWijaya, Sony Hartono, Farit Mochamad Afendi, Irmanida Batubara, Ming Huang, Naoaki Ono, Shigehiko Kanaya, and Md. Altaf-Ul-Amin. 2021. "Identification of Targeted Proteins by Jamu Formulas for Different Efficacies Using Machine Learning Approach" Life 11, no. 8: 866. https://doi.org/10.3390/life11080866
APA StyleWijaya, S. H., Afendi, F. M., Batubara, I., Huang, M., Ono, N., Kanaya, S., & Altaf-Ul-Amin, M. (2021). Identification of Targeted Proteins by Jamu Formulas for Different Efficacies Using Machine Learning Approach. Life, 11(8), 866. https://doi.org/10.3390/life11080866