
Identification of Compound CB-2 as a Novel Late-Stage Autophagy Inhibitor Exhibits Inhibitory Potency against A549 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Antibodies
2.3. Cell Culture and Treatment
2.4. Western Blotting (WB)
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Transmission Electron Microscopy (TEM)
2.7. Immunofluorescence and Confocal Microscopy
2.8. Real-Time Cell Analyzer Assay (RTCA)
2.9. Cell Viability Analysis
2.10. Annexin V-PITC/PI Double-Staining Analysis
2.11. Colony Formation Assay
2.12. LDH Assay
2.13. Wound-Healing Assay
2.14. Transwell Assay
2.15. Detection of Intracellular and Mitochondrial ROS Levels
2.16. Detection of Mitochondrial Membrane Potential (MMP)
2.17. Statistical Analysis
3. Results
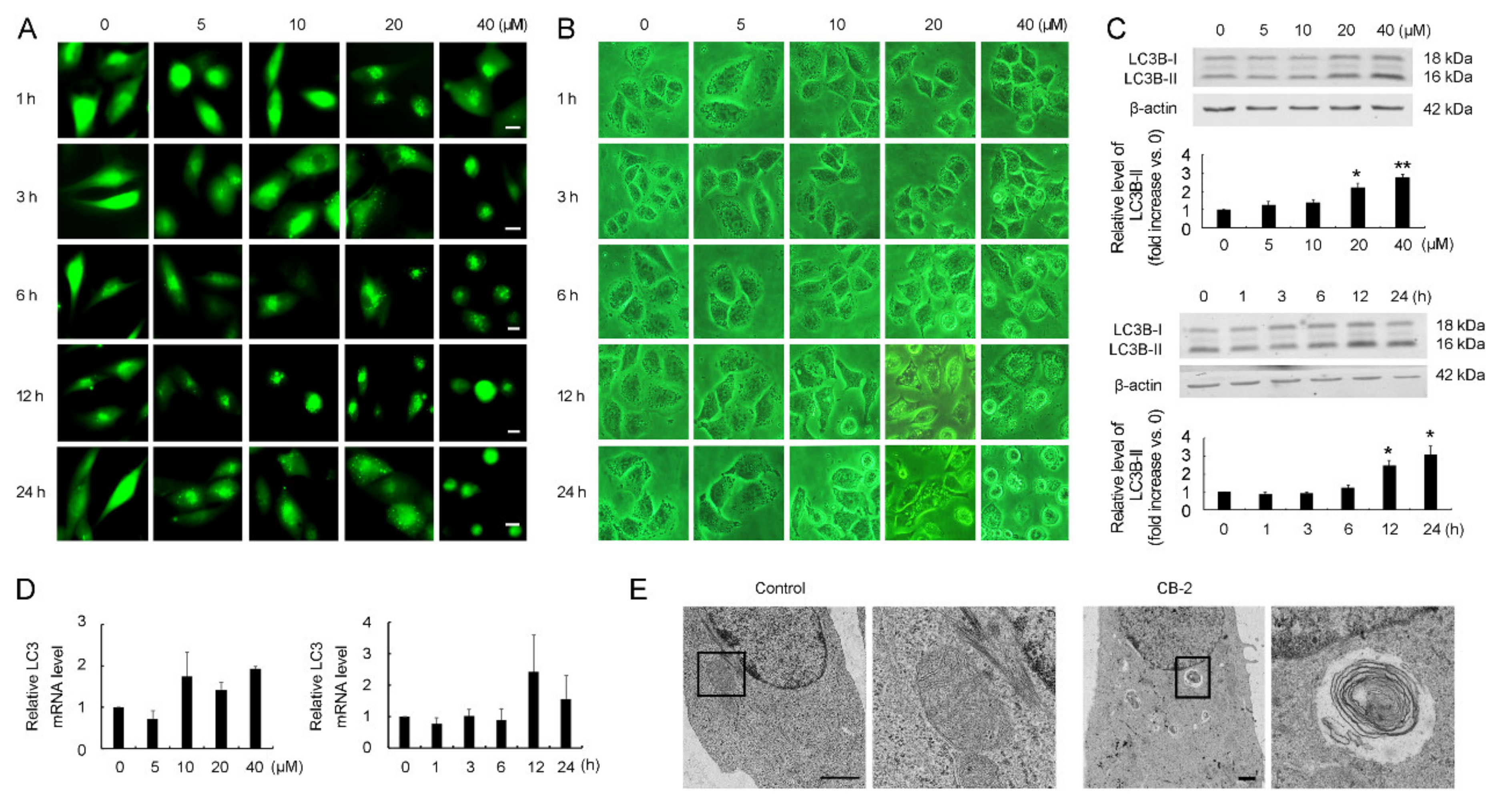
3.1. Discovery of CB-2 as an Autophagy Modulator
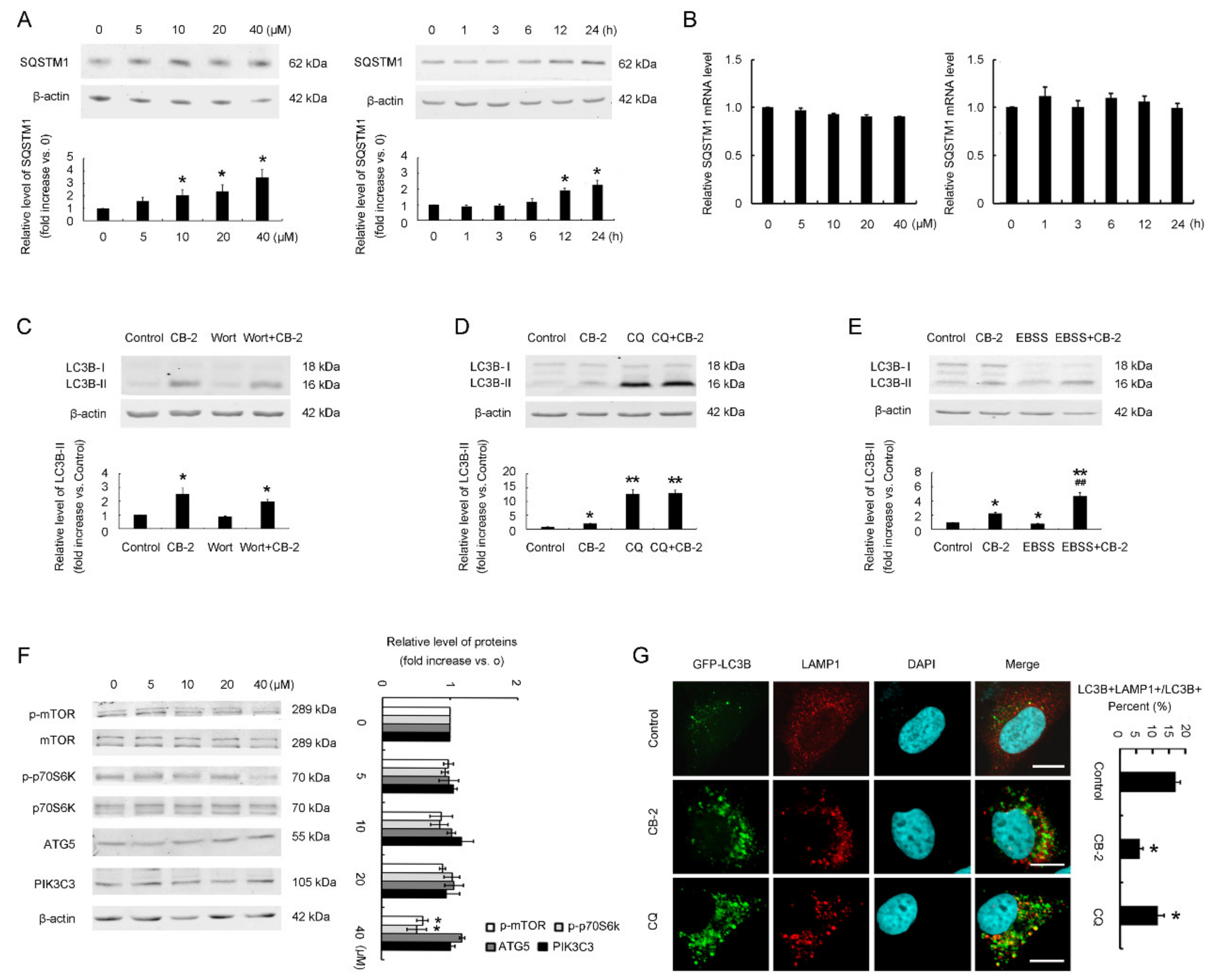
3.2. Identification of CB-2 as a Novel Inhibitor Targeting Autophagy at a Late Stage
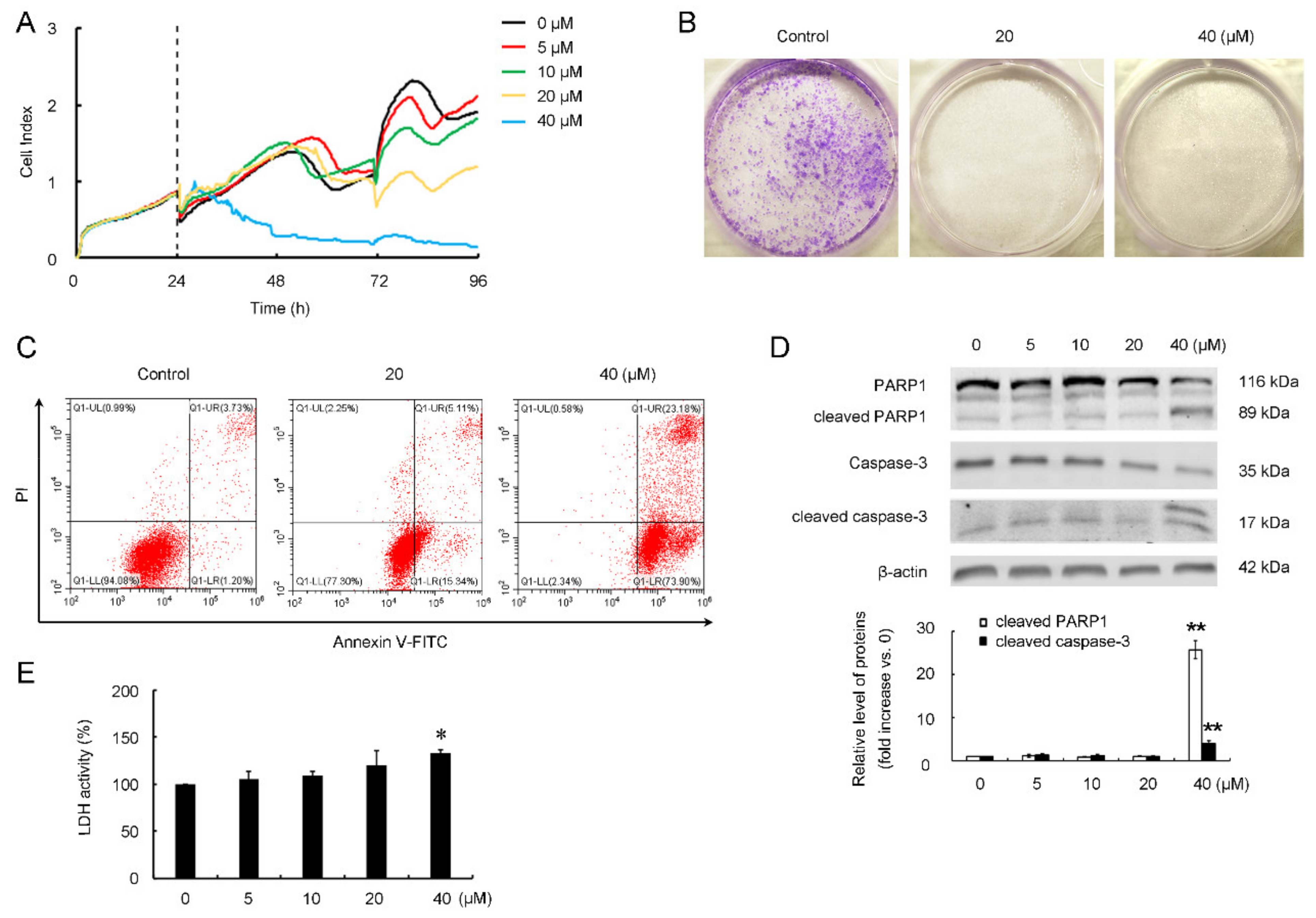
3.3. CB-2 Exhibits Inhibitory Activities against A549 Cells
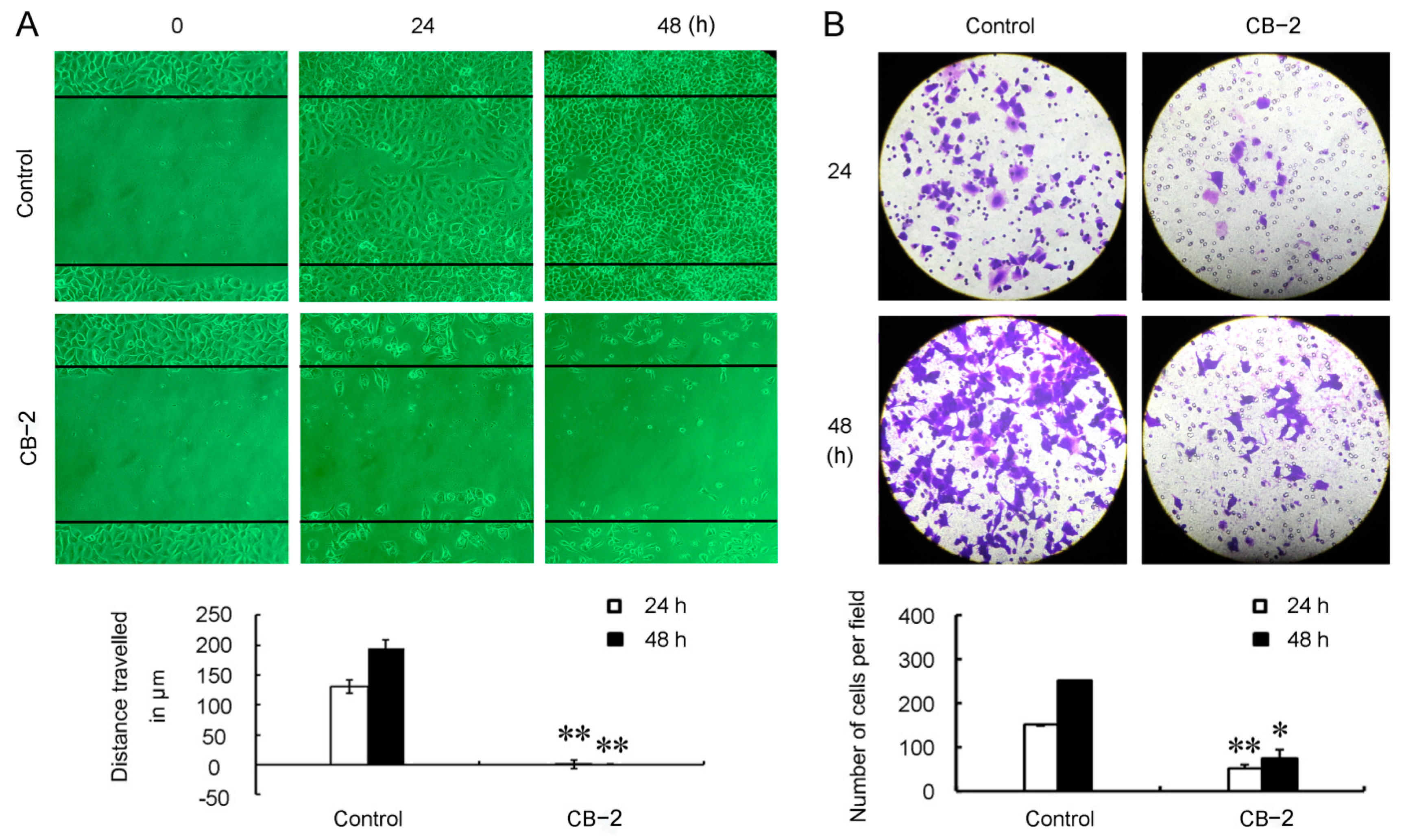
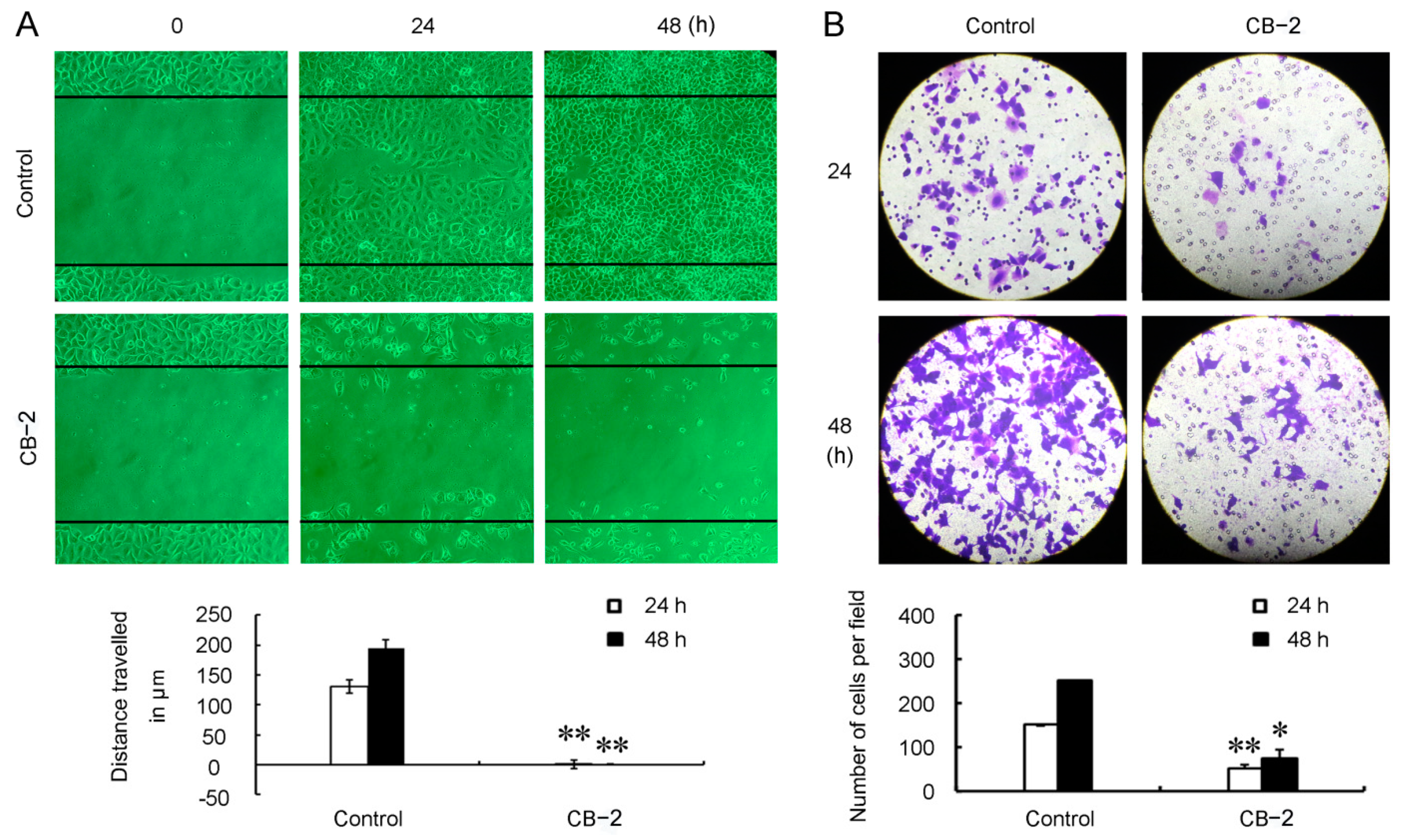
3.4. CB-2 Inhibits Migration of A549 Cells
3.5. CB-2 Induces Overproduction of Mitochondrial-Derived ROS and Decrease of MMP
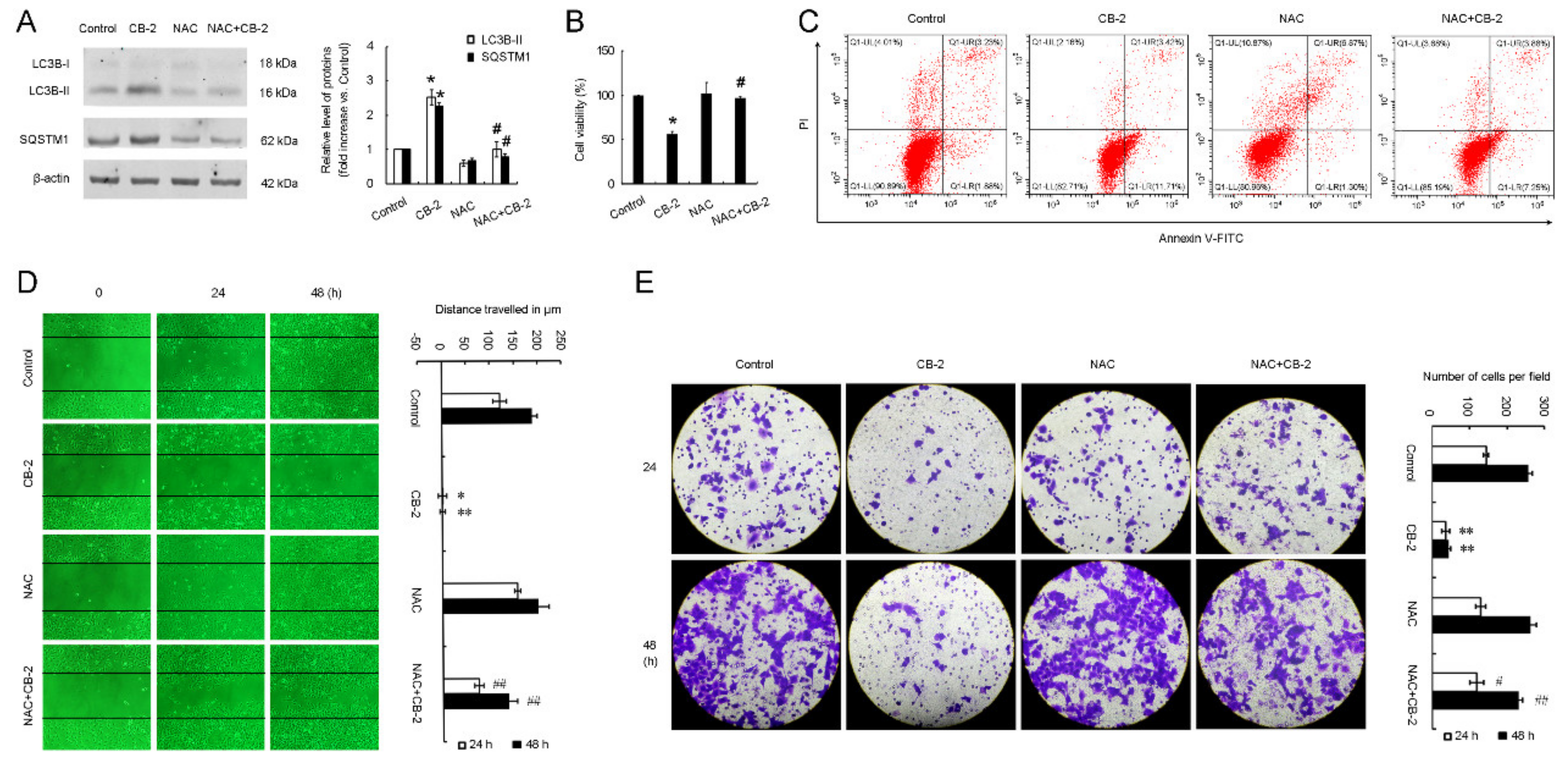
3.6. CB-2 Inhibits Autophagy and Exerts Inhibitory Potency against A549 Cells via ROS-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.; Emr, S. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy, metabolism, and cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and cancer dormancy. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2020, 1–9. [Google Scholar] [CrossRef]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy inhibition to augment mTOR inhibition: A phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.-T.; Yu, X.-X.; Yan, L.-J.; Xiao, H.-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Kawano, S.; Suto, T.; Sato, T.; Chida, N.; Simizu, S. Identification of madangamine A as a novel lysosomotropic agent to inhibit autophagy. Bioorganic Med. Chem. 2021, 34, 116041. [Google Scholar] [CrossRef]
- Zhang, L.; Qiang, P.; Yu, J.; Miao, Y.; Chen, Z.; Qu, J.; Zhao, Q.; Chen, Z.; Liu, Y.; Yao, X.; et al. Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer. Autophagy 2019, 15, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Deng, Q.; Zhang, H.; Hu, X.; Li, Y.; Liu, Y.; Hu, J.; Luo, Q.; Zhang, Y.; Jiang, X.; et al. A novel autophagy inhibitor berbamine blocks SNARE-mediated autophagosome-lysosome fusion through upregulation of BNIP3. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Zhao, X.; Fang, Y.; Yang, Y.; Qing, Y.; Wu, P.; Wang, T.; Lai, H.; Meng, L.; Wang, D.; Zheng, Z.; et al. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 2015, 11, 1849–1863. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Wan, G.; Liu, Z.; Wang, X.; Ruan, P.; Xu, W.; Xu, D.; Xie, W.; Zhang, Y.; Xu, H.; et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 2014, 10, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Gao, S.; Yang, Y.; Zhao, X.; Fan, Y.; Ma, W.; Yang, D.; Yang, A.; Yu, Y. Antitumor activity of curcumin by modulation of apoptosis and autophagy in human lung cancer A549 cells through inhibiting PI3K/Akt/mTOR pathway. Oncol. Rep. 2018, 39, 1523–1531. [Google Scholar] [CrossRef]
- Zhang, Q.; Qiao, H.; Wu, D.; Lu, H.; Liu, L.; Sang, X.; Li, D.; Zhou, Y. Curcumin potentiates the galbanic acid-induced anti-tumor effect in non-small cell lung cancer cells through inhibiting Akt/mTOR signaling pathway. Life Sci. 2019, 239, 117044. [Google Scholar] [CrossRef]
- Masuelli, L.; Benvenuto, M.; Di Stefano, E.; Mattera, R.; Fantini, M.; De Feudis, G.; De Smaele, E.; Tresoldi, I.; Giganti, M.G.; Modesti, A.; et al. Curcumin blocks autophagy and activates apoptosis of malignant mesothelioma cell lines and increases the survival of mice intraperitoneally transplanted with a malignant mesothelioma cell line. Oncotarget 2017, 8, 34405–34422. [Google Scholar] [CrossRef] [Green Version]
- Hewlings, S.; Kalman, D. Curcumin: A review of its effects on human health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Shabaninejad, Z.; Pourhanifeh, M.; Movahedpour, A.; Mottaghi, R.; Nickdasti, A.; Mortezapour, E.; Shafiee, A.; Hajighadimi, S.; Moradizarmehri, S.; Sadeghian, M.; et al. Therapeutic potential of curcumin in the treatment of glioblastoma multiforme. Eur. J. Med. Chem. 2020, 188, 112040. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, S.; Zhang, L.; Sun, G.; Chen, X. A synthetic curcumin derivative hydrazinobenzoylcurcumin induces autophagyin A549 lung cancer cells. Pharm. Biol. 2014, 52, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Yang, H.P.; Zhou, X.D.; Wang, H.J.; Gong, L.; Tang, C.L. Autophagy accompanied with bisdemethoxycurcumin-induced apoptosis in non-small cell lung cancer cells. Biomed. Environ. Sci. 2015, 28, 105–115. [Google Scholar] [CrossRef]
- Song, G.; Lu, H.; Chen, F.; Wang, Y.; Fan, W.; Shao, W.; Lu, H.; Lin, B. Tetrahydrocurcumin-induced autophagy via suppression of PI3K/Akt/mTOR in non-small cell lung carcinoma cells. Mol. Med. Rep. 2018, 17, 5964–5969. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dai, F.; Cui, L.; Jing, H.; Fan, P.; Tan, X.; Guo, Y.; Zhou, G.-Z. Novel role for TRPC4 in regulation of macroautophagy by a small molecule in vascular endothelial cells. Biochim. Biophys. Acta 2015, 1853, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.; Alekseev, B.Y.; Kardymon, O.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Mowers, E.E.; Sharifi, M.; MacLeod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, P.; Guo, F.; Wang, X.; Wang, J.; Xu, J.; Yuan, D.; Zhang, J.; Shao, C. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int. J. Radiat. Biol. 2017, 93, 764–770. [Google Scholar] [CrossRef]
- Salehi, M.; Movahedpour, A.; Tayarani, A.; Shabaninejad, Z.; Pourhanifeh, M.H.; Mortezapour, E.; Nickdasti, A.; Mottaghi, R.; Davoodabadi, A.; Khan, H.; et al. Therapeutic potentials of curcumin in the treatment of non-small-cell lung carcinoma. Phytother. Res. 2020, 34, 2557–2576. [Google Scholar] [CrossRef]
- Feng, C.; Xia, Y.; Zou, P.; Shen, M.; Hu, J.; Ying, S.; Pan, J.; Liu, Z.; Dai, X.; Zhuge, W.; et al. Curcumin analog L48H37 induces apoptosis through ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human lung cancer cells. Mol. Carcinog. 2017, 56, 1765–1777. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Y.; Liu, W.; Li, Z. T59, a new compound reconstructed from curcumin, induces cell apoptosis through reactive oxygen species activation in human lung cancer cells. Molecules 2018, 23, 1251. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhang, L.; Liu, Y.; Zhang, H.; Chen, J.; Feng, G.; Yang, P.; Sha, F.; Cui, L.; Sun, G. Identification of Compound CB-2 as a Novel Late-Stage Autophagy Inhibitor Exhibits Inhibitory Potency against A549 Cells. Life 2021, 11, 865. https://doi.org/10.3390/life11080865
Liu Z, Zhang L, Liu Y, Zhang H, Chen J, Feng G, Yang P, Sha F, Cui L, Sun G. Identification of Compound CB-2 as a Novel Late-Stage Autophagy Inhibitor Exhibits Inhibitory Potency against A549 Cells. Life. 2021; 11(8):865. https://doi.org/10.3390/life11080865
Chicago/Turabian StyleLiu, Zhihui, Lu Zhang, Yachao Liu, Hanxiao Zhang, Jingxuan Chen, Gaoqing Feng, Peichang Yang, Fangfang Sha, Liuqing Cui, and Gangchun Sun. 2021. "Identification of Compound CB-2 as a Novel Late-Stage Autophagy Inhibitor Exhibits Inhibitory Potency against A549 Cells" Life 11, no. 8: 865. https://doi.org/10.3390/life11080865
APA StyleLiu, Z., Zhang, L., Liu, Y., Zhang, H., Chen, J., Feng, G., Yang, P., Sha, F., Cui, L., & Sun, G. (2021). Identification of Compound CB-2 as a Novel Late-Stage Autophagy Inhibitor Exhibits Inhibitory Potency against A549 Cells. Life, 11(8), 865. https://doi.org/10.3390/life11080865