
Biological Hallmarks and New Therapeutic Approaches for the Treatment of PDAC


 ,
,
Abstract
:1. Introduction
2. KRAS Pathway
2.1. Mutational Status
2.2. KRAS Therapy
2.3. Clinical Trials
3. CDK4/6 Pathway
3.1. Mutational Status
3.2. CDK4/6 Therapy
4. Effect of Immunotherapy in Pancreatic Cancer
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Schnelldorfer, T.; Adams, D.B.; Warshaw, A.L.; Lillemoe, K.D.; Sarr, M.G. Forgotten pioneers of pancreatic surgery: Beyond the favorite few. Ann. Surg. 2008, 247, 191–202. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Scolnick, E.M.; Papageorge, A.G.; Shih, T.Y. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc. Natl. Acad. Sci. USA 1979, 76, 5355–5359. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Ardito, C.M.; Gruner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Velculescu, V.E.; Wolfgang, C.L.; Hruban, R.H. Genetic basis of pancreas cancer development and progression: Insights from whole-exome and whole-genome sequencing. Clin. Cancer Res. 2012, 18, 4257–4265. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; van der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.J.; Hancock, J.F.; et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albury, T.M.; Pandey, V.; Gitto, S.B.; Dominguez, L.; Spinel, L.P.; Talarchek, J.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A. Constitutively active Akt1 cooperates with KRas(G12D) to accelerate in vivo pancreatic tumor onset and progression. Neoplasia 2015, 17, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, Y.; Goldstein, J.L.; Seabra, M.C.; Casey, P.J.; Brown, M.S. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell 1990, 62, 81–88. [Google Scholar] [CrossRef]
- He, B.; Chen, P.; Chen, S.Y.; Vancura, K.L.; Michaelis, S.; Powers, S. RAM2, an essential gene of yeast, and RAM1 encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 11373–11377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.E.; Brunner, T.B.; Kiel, K.D.; DeLaney, T.F.; Regine, W.F.; Mohiuddin, M.; Rosato, E.F.; Haller, D.G.; Stevenson, J.P.; Smith, D.; et al. A phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin. Cancer Res. 2004, 10, 5447–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug. Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Taveras, A.G.; Remiszewski, S.W.; Doll, R.J.; Cesarz, D.; Huang, E.C.; Kirschmeier, P.; Pramanik, B.N.; Snow, M.E.; Wang, Y.S.; del Rosario, J.D.; et al. Ras oncoprotein inhibitors: The discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg. Med. Chem. 1997, 5, 125–133. [Google Scholar] [CrossRef]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [Green Version]
- Bao, G.Q.; Shen, B.Y.; Pan, C.P.; Zhang, Y.J.; Shi, M.M.; Peng, C.H. Andrographolide causes apoptosis via inactivation of STAT3 and Akt and potentiates antitumor activity of gemcitabine in pancreatic cancer. Toxicol. Lett. 2013, 222, 23–35. [Google Scholar] [CrossRef]
- Shu, Y.; Sun, J.; Cai, P.; Wang, W.; Han, X.; Gu, Y. An open-label, randomized, controlled clinical trial to explore the curative effects between the treatment of capecitabine and andrographolide and the single capecitabine in the patients with pathological and/or histologic diagnosed unresectable, advanced, recurrent, and metastatic colorectal cancer. J. Clin. Oncol. 2017, 35, TPS819. [Google Scholar]
- Robertson, K.D.; Jones, P.A. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene 1999, 18, 3810–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell. Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, G.; Fumarola, C.; La Monica, S.; Bonelli, M.A.; Cretella, D.; Alfieri, R.; Cavazzoni, A.; Galetti, M.; Bertolini, P.; Missale, G.; et al. Simultaneous Combination of the CDK4/6 Inhibitor Palbociclib With Regorafenib Induces Enhanced Anti-tumor Effects in Hepatocarcinoma Cell Lines. Front. Oncol. 2020, 10, 563249. [Google Scholar] [CrossRef] [PubMed]
- Edamoto, Y.; Hara, A.; Biernat, W.; Terracciano, L.; Cathomas, G.; Riehle, H.M.; Matsuda, M.; Fujii, H.; Scoazec, J.Y.; Ohgaki, H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int. J. Cancer 2003, 106, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Kern, S.E.; Hruban, R.H. Molecular pathogenesis of pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 211–226. [Google Scholar] [CrossRef]
- Knudsen, E.S.; O’Reilly, E.M.; Brody, J.R.; Witkiewicz, A.K. Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology 2016, 150, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Yang, P.; Liu, B.; Tang, Y. Is there a CDKN2A-centric network in pancreatic ductal adenocarcinoma? OncoTargets Ther. 2020, 13, 2551–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Liu, T.P.; Yang, P.M. CDKN2A-Inactivated Pancreatic Ductal Adenocarcinoma Exhibits Therapeutic Sensitivity to Paclitaxel: A Bioinformatics Study. J. Clin. Med. 2020, 9, 4019. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Jeong, J.; Park, Y.N.; Park, J.S.; Yoon, D.S.; Chi, H.S.; Kim, B.R. Clinical significance of p16 protein expression loss and aberrant p53 protein expression in pancreatic cancer. Yonsei Med. J. 2005, 46, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Fusaro, R.M. Pancreatic cancer and the familial atypical multiple mole melanoma (FAMMM) syndrome. Pancreas 1991, 6, 127–131. [Google Scholar] [CrossRef]
- De Snoo, F.A.; Bishop, D.T.; Bergman, W.; van Leeuwen, I.; van der Drift, C.; van Nieuwpoort, F.A.; Out-Luiting, C.J.; Vasen, H.F.; ter Huurne, J.A.; Frants, R.R.; et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin. Cancer Res. 2008, 14, 7151–7157. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, S.P.; Generali, D. Abemaciclib: A CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des. Dev. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A.; Tap, W.D.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Rathbone, D.D.; Condy, M.M.; et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [Google Scholar] [CrossRef] [Green Version]
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.H.; Xu, L.; Posey, J., 3rd; et al. Abemaciclib Is Effective Against Pancreatic Cancer Cells and Synergizes with HuR and YAP1 Inhibition. Mol. Cancer Res. 2019, 17, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Xu, Y.; Liu, B.; Singh, P.K.; Zhao, W.; Jin, J.; Han, G.; Scott, A.W.; Dong, X.; Huo, L.; et al. YAP1-Mediated CDK6 Activation Confers Radiation Resistance in Esophageal Cancer—Rationale for the Combination of YAP1 and CDK4/6 Inhibitors in Esophageal Cancer. Clin. Cancer Res. 2019, 25, 2264–2277. [Google Scholar] [CrossRef]
- Ziemke, E.K.; Dosch, J.S.; Maust, J.D.; Shettigar, A.; Sen, A.; Welling, T.H.; Hardiman, K.M.; Sebolt-Leopold, J.S. Sensitivity of KRAS-Mutant Colorectal Cancers to Combination Therapy That Cotargets MEK and CDK4/6. Clin. Cancer Res. 2016, 22, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Kumarasamy, V.; Chung, S.; Ruiz, A.; Vail, P.; Tzetzo, S.; Wu, J.; Nambiar, R.; Sivinski, J.; Chauhan, S.S.; et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut 2021, 70, 127–138. [Google Scholar] [CrossRef]
- Willobee, B.A.; Gaidarski, A.A.; Dosch, A.R.; Castellanos, J.A.; Dai, X.; Mehra, S.; Messaggio, F.; Srinivasan, S.; VanSaun, M.N.; Nagathihalli, N.S.; et al. Combined Blockade of MEK and CDK4/6 Pathways Induces Senescence to Improve Survival in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2018, 67, 2142–2155. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, D.; Jin, X.; Zhang, K. The CDK4/6 inhibitor PD0332991 stabilizes FBP1 by repressing MAGED1 expression in pancreatic ductal adenocarcinoma. Int. J. Biochem. Cell Biol. 2020, 128, 105859. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Shi, M.; Chen, H.; Gu, J.; Zhang, J.; Shen, B.; Deng, X.; Xie, J.; Zhan, X.; Peng, C. NPM1 activates metabolic changes by inhibiting FBP1 while promoting the tumorigenicity of pancreatic cancer cells. Oncotarget 2015, 6, 21443–21451. [Google Scholar] [CrossRef] [Green Version]
- Wattanavanitchakorn, S.; Rojvirat, P.; Chavalit, T.; MacDonald, M.J.; Jitrapakdee, S. CCAAT-enhancer binding protein-alpha (C/EBPalpha) and hepatocyte nuclear factor 4alpha (HNF4alpha) regulate expression of the human fructose-1,6-bisphosphatase 1 (FBP1) gene in human hepatocellular carcinoma HepG2 cells. PLoS ONE 2018, 13, e0194252. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, S.; Yang, H.; Deng, S.; Fan, P.; Li, M.; Jin, X. USP44 suppresses pancreatic cancer progression and overcomes gemcitabine resistance by deubiquitinating FBP1. Am. J. Cancer Res. 2019, 9, 1722–1733. [Google Scholar] [PubMed]
- Weon, J.L.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Pan, Y.; Wang, L.; Zhang, L.; Ravichandran, R.; Potts, P.R.; Jiang, J.; Wu, H.; Huang, H. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis 2017, 6, e312. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Borghaei, H.; Ramalingam, S.S.; Horn, L.; De Castro Carpeno, J.; Pluzanski, A.; Burgio, M.A.; Garassino, M.; Chow, L.Q.M.; Gettinger, S.; et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: A pooled analysis. Lancet Oncol. 2019, 20, 1395–1408. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Johnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and local immunosuppression in pancreatic cancer patients. Clin. Cancer Res. 2001, 7, 925S–932S. [Google Scholar]
- Beatty, G.L.; Eghbali, S.; Kim, R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grunwald, V.; Hutson, T.E.; Kopyltsov, E.; Mendez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Principe, D.R.; Korc, M.; Kamath, S.D.; Munshi, H.G.; Rana, A. Trials and tribulations of pancreatic cancer immunotherapy. Cancer Lett. 2021, 504, 1–14. [Google Scholar] [CrossRef]
- Shibuya, K.C.; Goel, V.K.; Xiong, W.; Sham, J.G.; Pollack, S.M.; Leahy, A.M.; Whiting, S.H.; Yeh, M.M.; Yee, C.; Riddell, S.R.; et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS ONE 2014, 9, e96565. [Google Scholar] [CrossRef]
- Homma, Y.; Taniguchi, K.; Murakami, T.; Nakagawa, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ueda, M.; Ichikawa, Y.; et al. Immunological impact of neoadjuvant chemoradiotherapy in patients with borderline resectable pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 670–676. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbaek, M.S.; Simoes, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, R.; Giovannetti, E.; Bonelli, M.; Cavazzoni, A. New Treatment Opportunities in Phosphatase and Tensin Homolog (PTEN)-Deficient Tumors: Focus on PTEN/Focal Adhesion Kinase Pathway. Front. Oncol. 2017, 7, 170. [Google Scholar] [CrossRef]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef] [Green Version]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions With Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Le Large, T.Y.S.; Bijlsma, M.F.; El Hassouni, B.; Mantini, G.; Lagerweij, T.; Henneman, A.A.; Funel, N.; Kok, B.; Pham, T.V.; de Haas, R.; et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 91. [Google Scholar] [CrossRef]
- Osipov, A.; Blair, A.B.; Liberto, J.; Wang, J.; Li, K.; Herbst, B.; Xu, Y.; Li, S.; Niu, N.; Rashid, R.; et al. Inhibition of focal adhesion kinase enhances antitumor response of radiation therapy in pancreatic cancer through CD8+ T cells. Cancer Biol. Med. 2021, 18, 206–214. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Thomsen, A.; Follo, M.; Zamboglou, C.; Bronsert, P.; Mostafa, H.; Amen, A.; Mekawy, M.; Grosu, A.L.; Brunner, T.B. FAK inhibition radiosensitizes pancreatic ductal adenocarcinoma cells in vitro. Strahlenther. Onkol. 2021, 197, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, S.N.; Anderton, S.M.; Serrels, A. FAK-inhibition opens the door to checkpoint immunotherapy in Pancreatic Cancer. J. Immunother. Cancer 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidaway, P. Pancreatic cancer: FAK regulates sensitivity to immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Sun, Z.; Ren, Z.; Yang, K.; Liu, Z.; Cao, S.; Deng, S.; Xu, L.; Liang, Y.; Guo, J.; Bian, Y.; et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8(+) T-cell response and effective tumor control. Nat. Commun. 2019, 10, 3874. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, Z.; Du, X.; Chen, S.; Zhang, W.; Wang, J.; Li, H.; He, X.; Cao, J.; Wang, J. Co-inhibition of the TGF-beta pathway and the PD-L1 checkpoint by pH-responsive clustered nanoparticles for pancreatic cancer microenvironment regulation and anti-tumor immunotherapy. Biomater. Sci. 2020, 8, 5121–5132. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing effective combination therapy for pancreatic cancer: An overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, T.P.; Vahrmeijer, A.L.; de Miranda, N. Immunotherapy for pancreatic cancer: Chasing the light at the end of the tunnel. Cell Oncol. 2021, 44, 261–278. [Google Scholar] [CrossRef] [PubMed]
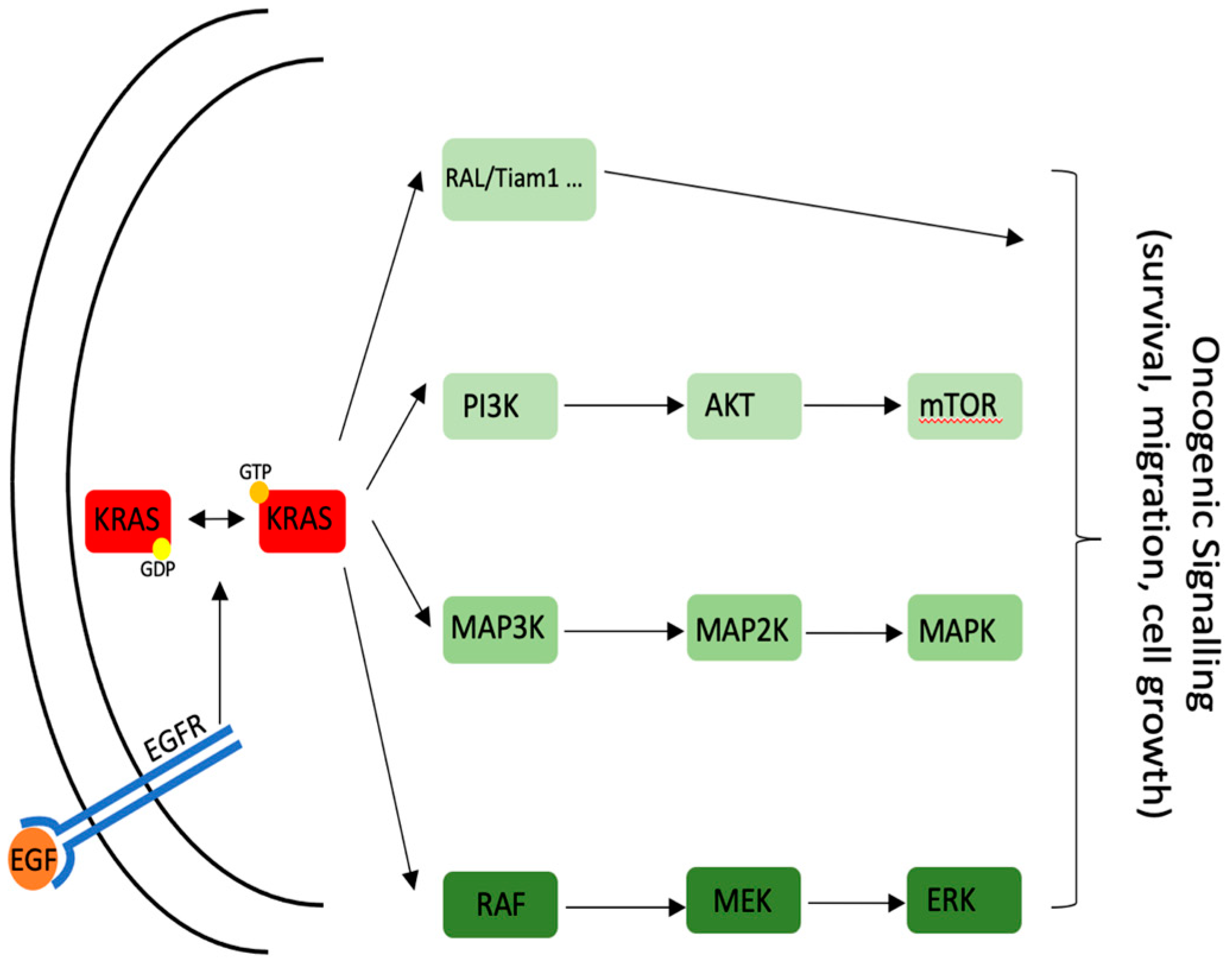

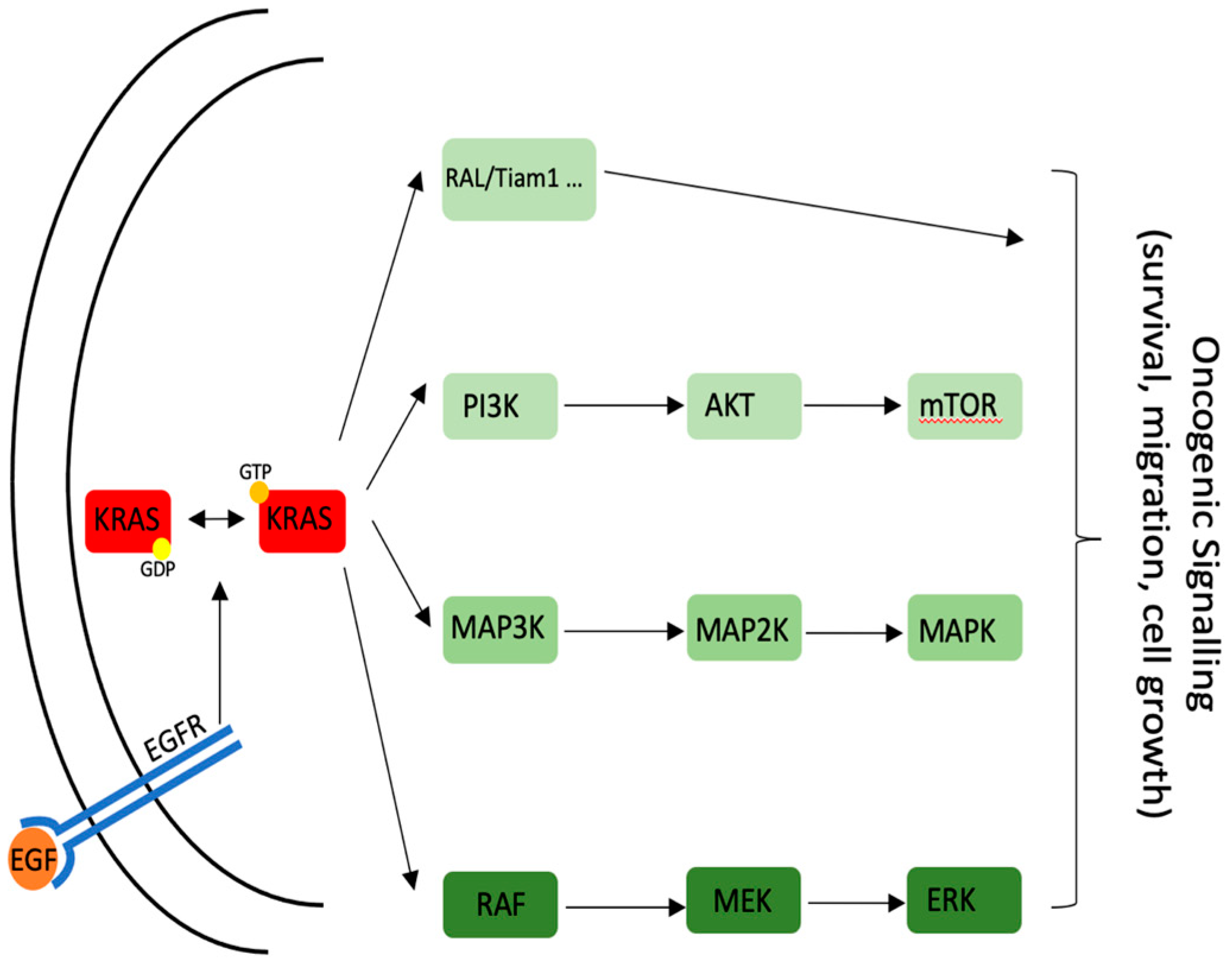{kind=link}
| Agent | Target | Combination | Phase | Reference |
|---|---|---|---|---|
| MRTX849 | KRAS G12C | - | I/II | NCT03785249 |
| Exosome G12D siRNA | KRAS G12D | - | I | NCT03608631 |
| NBF-006 | KRAS | - | I | NCT03819387 |
| Agent | Target | Combination | Phase | References |
|---|---|---|---|---|
| Palbociclib | CDK4/6 | Ulixertinib | I | NCT03454035 |
| Palbociclib | CDK4/6 | Gedatolisib | I | NCT03065062 |
| Palbociclib | CDK4/6 | Carboplatin Cisplatin | I | NCT02897375 |
| Palbociclib | CDK4/6 | II | NCT02806648 | |
| Palbociclib | CDK4/6 | II | NCT02465060 | |
| Abemaciclib Palbociclib | CDK4/6 | I | NCT03878524 | |
| Abemaciclib | CDK4/6 | LY3023414 Gemcitabine Capecitabine | II | NCT02981342 |
| Abemaciclib | CDK4/6 | II | NCT03891784 | |
| Abemaciclib | CDK4/6 | LY3300054 | I | NCT02791334 |
| Ribociclib | CDK4/6 | Trametinib | I/II | NCT02703571 |
| Ribociclib | CDK4/6 | II | NCT02420691 |
| Agent | Combination | Phase | Reference |
|---|---|---|---|
| FOLFIRINOX | Anti PD-1 | III | NCT03977272 |
| Gemcitabine or FOLFIRINOX | Pembrolizumab | II | NCT04447092 |
| Gemcitabine or S-1 | Nivolumab | II | NCT04377048 |
| FOLFIRINOX | BsAb PD-1/CTLA-4 | I/II | NCT04324307 |
| Gemcitabine/nab-paclitaxel | Nivolumab/Ipilimumab | I/II | NCT04247165 |
| FOLFIRINOX | Nivolumab | I/II | NCT03970252 |
| FOLFIRINOX | Anti PD-1 | III | NCT03983057 |
| Gemcitabine/nab-paclitaxel | Camrelizumab | III | NCT04674956 |
| Agent | Target | Combination | Phase | Reference |
|---|---|---|---|---|
| Anlotinib (AK105) | Multi-kinase | Anti-PD-1 | I/II | NCT04803851 |
| Defactinib | FAK | Pembrolizumab | I/II | NCT02758587 |
| LYT-200 | Galectin-9 | Anti PD-1 | I/II | NCT04666688 |
| Anetumab Ravtansine | Mesothelin | Nivolumab Nivolumab/Ipilimumab Nivolumab/gemcitabine | I/II | NCT03816358 |
| Anlotinib | Multi-kinase | Toripalimab | I/II | NCT04718701 |
| Olaptesed pegol (NOX-A12) | CXCL12 | Pembrolizumab | I/II | NCT03168139 |
| Entinostat | HDAC | Nivolumab | II | NCT03250273 |
| Galunisertib | TGFβRI | Durvalumab | I | NCT02734160 |
| Merestinib | MET | LY3300054 | I | NCT02791334 |
| Pexidartinib | CSF1R | Durvalumab | I | NCT02777710 |
| Danvatirsen | STAT3 | Durvalumab | II | NCT02983578 |
| Plerixafor | Hematopoietic stem cells | Cemiplimab | II | NCT04177810 |
| KY1044 | ICOS | Atezolizumab | I/II | NCT03829501 |
| Ibrutinib | BTK | Durvalumab | I/II | NCT02403271 |
| Defactinib | FAK | Pembrolizumab | II | NCT03727880 |
| Itacitinib | JAK1 | Pembrolizumab | I | NCT02646748 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Digiacomo, G.; Volta, F.; Garajova, I.; Balsano, R.; Cavazzoni, A. Biological Hallmarks and New Therapeutic Approaches for the Treatment of PDAC. Life 2021, 11, 843. https://doi.org/10.3390/life11080843
Digiacomo G, Volta F, Garajova I, Balsano R, Cavazzoni A. Biological Hallmarks and New Therapeutic Approaches for the Treatment of PDAC. Life. 2021; 11(8):843. https://doi.org/10.3390/life11080843
Chicago/Turabian StyleDigiacomo, Graziana, Francesco Volta, Ingrid Garajova, Rita Balsano, and Andrea Cavazzoni. 2021. "Biological Hallmarks and New Therapeutic Approaches for the Treatment of PDAC" Life 11, no. 8: 843. https://doi.org/10.3390/life11080843
APA StyleDigiacomo, G., Volta, F., Garajova, I., Balsano, R., & Cavazzoni, A. (2021). Biological Hallmarks and New Therapeutic Approaches for the Treatment of PDAC. Life, 11(8), 843. https://doi.org/10.3390/life11080843