
Platelet APP Processing: Is It a Tool to Explore the Pathophysiology of Alzheimer’s Disease? A Systematic Review

 ,
,  and
and 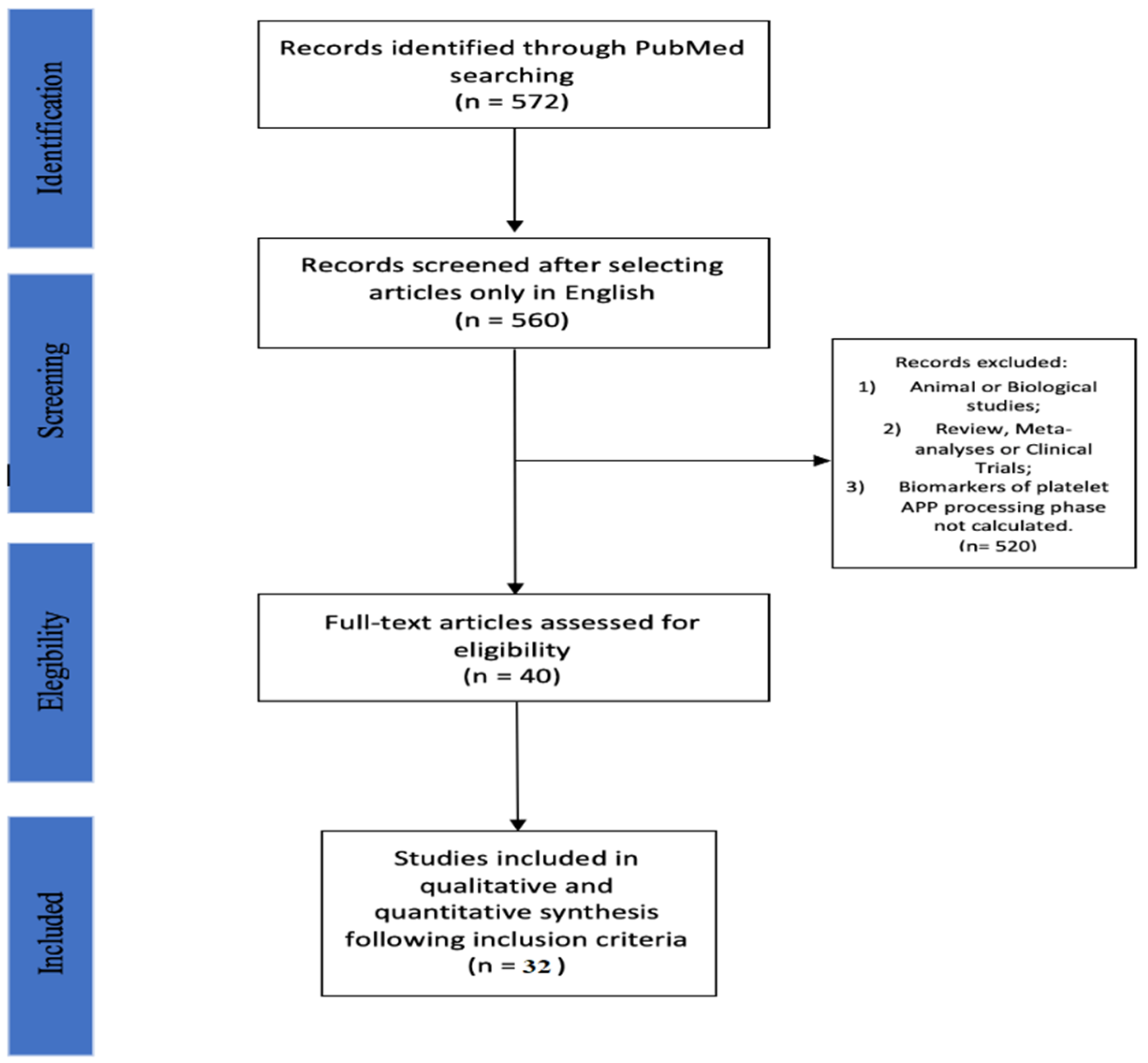{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Alzheimer’s Disease
1.2. The Diagnosis Limits
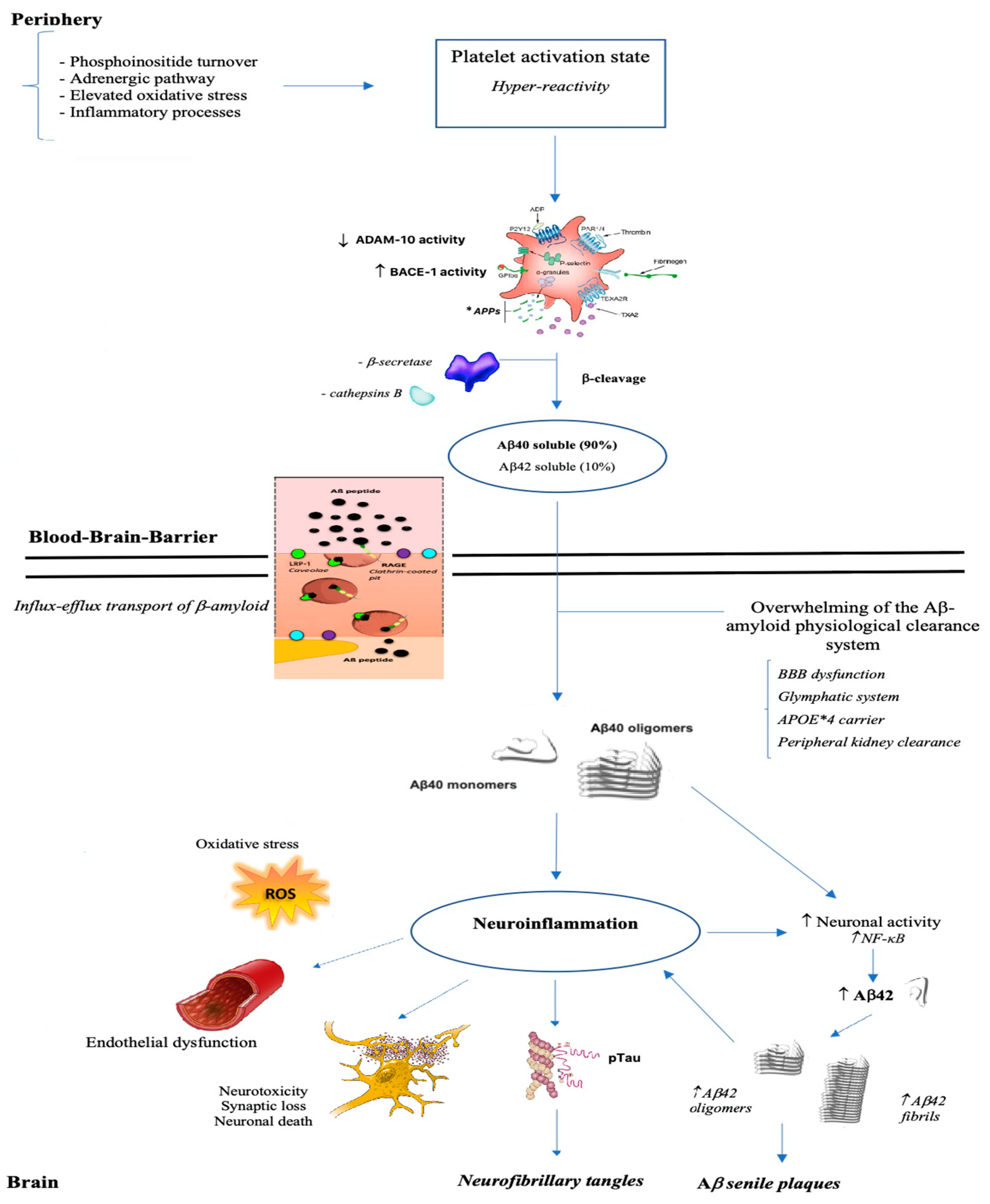
1.3. The APP Processing Phase
1.4. Platelets and AD
2. Materials and Methods
2.1. Search Strategy
2.2. Selection Criteria
2.3. Data Extraction
2.4. Quality Assessment
- A paper with no weak rating is considered Strong, in turn divided into Very Strong (6) if 2 ≤ Moderate ratings are present and Strong (5) if 2 > Moderate rating are present;
- A paper with 1 weak rating is considered Moderate, in turn divided into Very Moderate (4) if there are 2 ≤ Moderate ratings and Moderate (3) if there are 2 > Moderate rating;
- A paper with 2 weak ratings is considered Weak, in turn divided into Very Weak (2) if there are 2 ≤ Moderate ratings and Weak (1) if there are 2 > Moderate rating or >3 of Weak ratings.
3. Results
3.1. Studies Analyzing APP Ratio
3.2. Studies Detecting ADAM-10 and/or BACE-1 Activities
3.3. Study Calculating Platelet Aβ40/42
3.4. Calculation of mRNA-APP Isoform
3.5. APP Isoforms Expressed as N-Terminal and C-Terminal
3.6. Platelet PSEN-1 Activity
3.7. Differences in the Expression of APP Isoforms
3.8. APP Expressed on Platelet Surface and sAPP Released by Platelets
4. Discussion
4.1. Amyloid Protein Precursor (APP)
4.2. The APP Processing System
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kirson, N.Y.; Desai, U.; Ristovska, L.; Cummings, A.K.; Birnbaum, H.G.; Ye, W.; Andrews, J.S.; Ball, D.; Kahle-Wrobleski, K. Assessing the economic burden of Alzheimer’s disease patients first diagnosed by specialists. BMC Geriatr. 2016, 16, 138. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H. Increasing burden of Alzheimer’s disease by aging. J. Korean Med. Sci. 2014, 29, 885. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Kljajevic, V. Overestimating the Effects of Healthy Aging. Front. Aging Neurosci. 2015, 7, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Lowe, V.J.; Weigand, S.D.; Wiste, H.J.; Senjem, M.L.; Knopman, D.S.; Shiung, M.M.; Gunter, J.L.; Boeve, B.F.; Kemp, B.J.; et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: Implications for sequence of pathological events in Alzheimer’s disease. Brain 2009, 132, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Dominantly Inherited Alzheimer, N., Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Del Tredici, K.; Ludolph, A.C.; Hoozemans, J.J.; Rozemuller, A.J.; Braak, H.; Knippschild, U. Stages of granulovacuolar degeneration: Their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 2011, 122, 577–589. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Muller, U.C.; Deller, T. Editorial: The Physiological Functions of the APP Gene Family. Front Mol. Neurosci. 2017, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Muller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Holtzman, D.M. Role of apoe/Abeta interactions in the pathogenesis of Alzheimer’s disease and cerebral amyloid angiopathy. J. Mol. Neurosci. 2001, 17, 147–155. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Greenberg, S.M. Beta-amyloid, blood vessels, and brain function. Stroke 2009, 40, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, L.Y.; Davila-Rodriguez, J.; Rivera-Aponte, D.E.; Zueva, L.V.; Washington, A.V.; Sanabria, P.; Inyushin, M.Y. Platelets are responsible for the accumulation of beta-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res. Bull 2017, 128, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Li, Q.X. Platelets and Alzheimer’s disease: Potential of APP as a biomarker. World J. Psychiatry 2012, 2, 102–113. [Google Scholar] [CrossRef]
- Dong, Y.; Lagarde, J.; Xicota, L.; Corne, H.; Chantran, Y.; Chaigneau, T.; Crestani, B.; Bottlaender, M.; Potier, M.C.; Aucouturier, P.; et al. Neutrophil hyperactivation correlates with Alzheimer’s disease progression. Ann. Neurol. 2018, 83, 387–405. [Google Scholar] [CrossRef] [PubMed]
- Rossor, M.N.; Fox, N.C.; Beck, J.; Campbell, T.C.; Collinge, J. Incomplete penetrance of familial Alzheimer’s disease in a pedigree with a novel presenilin-1 gene mutation. Lancet 1996, 347, 1560. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Oltra-Cucarella, J.; Ferrer-Cascales, R.; Alegret, M.; Gasparini, R.; Diaz-Ortiz, L.M.; Rios, R.; Martinez-Nogueras, A.L.; Onandia, I.; Perez-Vicente, J.A.; Cabello-Rodriguez, L.; et al. Risk of progression to Alzheimer’s disease for different neuropsychological Mild Cognitive Impairment subtypes: A hierarchical meta-analysis of longitudinal studies. Psychol. Aging 2018, 33, 1007–1021. [Google Scholar] [CrossRef]
- Jost, B.C.; Grossberg, G.T. The natural history of Alzheimer’s disease: A brain bank study. J. Am. Geriatr. Soc. 1995, 43, 1248–1255. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Rowe, C.C. Amyloid imaging. Int. Psychogeriatr. 2011, 23 (Suppl. 2), S41–S49. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Ong, K.; Mulligan, R.S.; Holl, G.; Pejoska, S.; Jones, G.; O’Keefe, G.; Ackerman, U.; Tochon-Danguy, H.; Chan, J.G.; et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 2011, 52, 1210–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villemagne, V.L.; O’Keefe, G.; Mulligan, R.S.; Rowe, C.C. Quantitative approaches to amyloid imaging. Methods Mol. Biol. 2011, 680, 201–225. [Google Scholar]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Buerger, K.; Zinkowski, R.; Teipel, S.J.; Goernitz, A.; Andreasen, N.; Sjoegren, M.; DeBernardis, J.; Kerkman, D.; Ishiguro, K.; et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: A comparative cerebrospinal fluid study. Arch. Gen. Psychiatry 2004, 61, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattsson, N.; Zetterberg, H.; Hansson, O.; Andreasen, N.; Parnetti, L.; Jonsson, M.; Herukka, S.K.; van der Flier, W.M.; Blankenstein, M.A.; Ewers, M.; et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009, 302, 385–393. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H. Future screening for incipient Alzheimer’s disease--the influence of prevalence on test performance. Eur. Neurol. 2009, 62, 200–203. [Google Scholar] [CrossRef]
- Blennow, K. A Review of Fluid Biomarkers for Alzheimer’s Disease: Moving from CSF to Blood. Neurology 2017, 6 (Suppl. 1), 15–24. [Google Scholar] [CrossRef]
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lonneborg, A.; Wyss-Coray, T.; Soares, H.; et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 115–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galasko, D.; Golde, T.E. Biomarkers for Alzheimer’s disease in plasma, serum and blood—Conceptual and practical problems. Alzheimers Res. 2013, 5, 10. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Ohrfelt, A.; Portelius, E.; Bjerke, M.; Holtta, M.; Rosen, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Chen, M.; Inestrosa, N.C.; Ross, G.S.; Fernandez, H.L. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem. Biophys. Res. Commun. 1995, 213, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.L. Platelet secretory mechanisms. Semin. Thromb. Hemost. 2004, 30, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C. Stimulated release of the beta-amyloid protein of Alzheimer’s disease by normal human platelets. Neurosci. Lett. 1997, 235, 157–159. [Google Scholar] [CrossRef]
- Milovanovic, M.; Eriksson, K.; Winblad, B.; Nilsson, S.; Lindahl, T.L.; Post, C.; Jaremo, P. Alzheimer and platelets: Low-density platelet populations reveal increased serotonin content in Alzheimer type dementia. Clin. Biochem. 2014, 47, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Rainesalo, S.; Keranen, T.; Saransaari, P.; Honkaniemi, J. GABA and glutamate transporters are expressed in human platelets. Brain Res. Mol. Brain Res. 2005, 141, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Wojsiat, J.; Laskowska-Kaszub, K.; Mietelska-Porowska, A.; Wojda, U. Search for Alzheimer’s disease biomarkers in blood cells: Hypotheses-driven approach. Biomark. Med. 2017, 11, 917–931. [Google Scholar] [CrossRef] [Green Version]
- Inyushin, M.Y.; Sanabria, P.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Abeta Peptide Originated from Platelets Promises New Strategy in Anti-Alzheimer’s Drug Development. Biomed. Res. Int. 2017, 2017, 3948360. [Google Scholar] [CrossRef]
- Gasecka, A.; Boing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2017, 28, 228–234. [Google Scholar] [CrossRef]
- Di Luca, M.C.F.; Pastorino, L.; Borroni, B.; Padovani, A.; Cattabeni, F. Platelets as a peripheral district where to study pathogenetic mechanisms of alzheimer disease: The case of amyloid precursor protein. Eur. J. Pharm. 2000, 405, 277–283. [Google Scholar] [CrossRef]
- Bayer, T.A.; Cappai, R.; Masters, C.L.; Beyreuther, K.; Multhaup, G. It all sticks together—The APP-related family of proteins and Alzheimer’s disease. Mol. Psychiatry 1999, 4, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovronsky, D.M.; Lee, V.M.; Pratico, D. Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J. Biol. Chem. 2001, 276, 17036–17043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racchi, M.; Govoni, S. The pharmacology of amyloid precursor protein processing. Exp. Gerontol. 2003, 38, 145–157. [Google Scholar] [CrossRef]
- Haass, C. Take five--BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Zainaghi, I.A.; Forlenza, O.V.; Gattaz, W.F. Abnormal APP processing in platelets of patients with Alzheimer’s disease: Correlations with membrane fluidity and cognitive decline. Psychopharmacology 2007, 192, 547–553. [Google Scholar] [CrossRef]
- Muller, U.C.; Pietrzik, C.U.; Deller, T. The physiological functions of the beta-amyloid precursor protein APP. Exp. Brain Res. 2012, 217, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Visconte, C.; Canino, J.; Guidetti, G.F.; Zara, M.; Seppi, C.; Abubaker, A.A.; Pula, G.; Torti, M.; Canobbio, I. Amyloid precursor protein is required for in vitro platelet adhesion to amyloid peptides and potentiation of thrombus formation. Cell Signal 2018, 52, 95–102. [Google Scholar] [CrossRef]
- Silva, J.V.; Yoon, S.; Domingues, S.; Guimaraes, S.; Goltsev, A.V.; da Cruz, E.S.E.F.; Mendes, J.F.; da Cruz, E.S.O.A.; Fardilha, M. Amyloid precursor protein interaction network in human testis: Sentinel proteins for male reproduction. BMC Bioinform. 2015, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Sloane, J.A.; Pietropaolo, M.F.; Rosene, D.L.; Moss, M.B.; Peters, A.; Kemper, T.; Abraham, C.R. Lack of correlation between plaque burden and cognition in the aged monkey. Acta Neuropathol. 1997, 94, 471–478. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-beta Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [PubMed] [Green Version]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monning, U.; Konig, G.; Banati, R.B.; Mechler, H.; Czech, C.; Gehrmann, J.; Schreiter-Gasser, U.; Masters, C.L.; Beyreuther, K. Alzheimer beta A4-amyloid protein precursor in immunocompetent cells. J. Biol. Chem. 1992, 267, 23950–23956. [Google Scholar] [CrossRef]
- Golde, T.E.; Estus, S.; Usiak, M.; Younkin, L.H.; Younkin, S.G. Expression of beta amyloid protein precursor mRNAs: Recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron 1990, 4, 253–267. [Google Scholar] [CrossRef]
- Krieger, M.A.; Salles, J.M.; Almeida, E.; Linss, J.; Bonaldo, M.C.; Goldenberg, S. Expression and polymorphism of a Trypanosoma cruzi gene encoding a cytoplasmic repetitive antigen. Exp. Parasitol. 1990, 70, 247–254. [Google Scholar] [CrossRef]
- Li, Q.X.; Berndt, M.C.; Bush, A.I.; Rumble, B.; Mackenzie, I.; Friedhuber, A.; Beyreuther, K.; Masters, C.L. Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer’s disease in human platelet and brain: Surface expression on the activated human platelet. Blood 1994, 84, 133–142. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.D.; Liu, K.N.; Luo, Y.; Slack, J.L.; Stocking, K.L.; Peschon, J.J.; Johnson, R.S.; Castner, B.J.; Cerretti, D.P.; Black, R.A. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 1998, 273, 27765–27767. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. BACE1 structure and function in health and Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 100–120. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingtdeux, V.; Marambaud, P. Identification and biology of alpha-secretase. J. Neurochem. 2012, 120 (Suppl. 1), 34–45. [Google Scholar] [CrossRef] [PubMed]
- Sobol, A.; Galluzzo, P.; Liang, S.; Rambo, B.; Skucha, S.; Weber, M.J.; Alani, S.; Bocchetta, M. Amyloid precursor protein (APP) affects global protein synthesis in dividing human cells. J. Cell Physiol. 2015, 230, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Gandy, S. Lifelong management of amyloid-beta metabolism to prevent Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, C.; Demos, C.M.; Rao, K.S.; Pappolla, M.A.; Sambamurti, K. Beta-secretase: Structure, function, and evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.; Ghiso, J.; Wisniewski, T.; Frangione, B. Alzheimer’s presenilin 1 gene expression in platelets and megakaryocytes. Identification of a novel splice variant. FEBS Lett. 1996, 393, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganjei, J.K. Targeting amyloid precursor protein secretases: Alzheimer’s disease and beyond. Drug News Perspect. 2010, 23, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Wadhwa, P.K.; Rohilla, S.; Jadhav, H.R. Rational design, synthesis and in vitro evaluation of allylidene hydrazinecarboximidamide derivatives as BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, N.; Shoji, M.; Arai, H.; Furukawa, K.; Ikeuchi, T.; Akazawa, K.; Takami, M.; Hatsuta, H.; Murayama, S.; Hashimoto, Y.; et al. Japanese Alzheimer’s Disease Neuroimaging, I., Altered gamma-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO Mol. Med. 2012, 4, 344–352. [Google Scholar] [CrossRef]
- Takami, M.; Funamoto, S. gamma-Secretase-Dependent Proteolysis of Transmembrane Domain of Amyloid Precursor Protein: Successive Tri- and Tetrapeptide Release in Amyloid beta-Protein Production. Int. J. Alzheimers Dis. 2012, 2012, 591392. [Google Scholar] [PubMed] [Green Version]
- Smirnov, A.; Trupp, A.; Henkel, A.W.; Bloch, E.; Reulbach, U.; Lewczuk, P.; Riggert, J.; Kornhuber, J.; Wiltfang, J. Differential processing and secretion of Abeta peptides and sAPPalpha in human platelets is regulated by thrombin and prostaglandine 2. Neurobiol. Aging 2009, 30, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Marcello, E.; Borroni, B.; Kamphuis, W.; Hol, E.; Padovani, A.; Nascimento, C.C.; de Godoy Bueno, P.; Assis Carvalho Vale, F.; Iost Pavarini, S.C.; et al. ADAM10 gene expression in the blood cells of Alzheimer’s disease patients and mild cognitive impairment subjects. Biomarkers 2015, 20, 196–201. [Google Scholar] [CrossRef]
- Vignini, A.; Sartini, D.; Morganti, S.; Nanetti, L.; Luzzi, S.; Provinciali, L.; Mazzanti, L.; Emanuelli, M. Platelet amyloid precursor protein isoform expression in Alzheimer’s disease: Evidence for peripheral marker. Int. J. Immunopathol. Pharm. 2011, 24, 529–534. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 2004, 256, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C. Challenges of epidemiological studies of mild cognitive impairment. Alzheimer Dis. Assoc. Disord. 2004, 18, 1–2. [Google Scholar] [CrossRef]
- Manzine, P.R.; de Franca Bram, J.M.; Barham, E.J.; do Vale Fde, A.; Selistre-de-Araujo, H.S.; Cominetti, M.R.; Iost Pavarini, S.C. ADAM10 as a biomarker for Alzheimer’s disease: A study with Brazilian elderly. Dement Geriatr. Cogn. Disord. 2013, 35, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Marcello, E.; Borroni, B.; Zimmermann, M.; Caltagirone, C.; Cattabeni, F.; Padovani, A.; Di Luca, M. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology 2004, 62, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Bram, J.M.F.; Talib, L.L.; Joaquim, H.P.G.; Sarno, T.A.; Gattaz, W.F.; Forlenza, O.V. Protein levels of ADAM10, BACE1, and PSEN1 in platelets and leukocytes of Alzheimer’s disease patients. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [Green Version]
- Armijo-Olivo, S.; Stiles, C.R.; Hagen, N.A.; Biondo, P.D.; Cummings, G.G. Assessment of study quality for systematic reviews: A comparison of the Cochrane Collaboration Risk of Bias Tool and the Effective Public Health Practice Project Quality Assessment Tool: Methodological research. J. Eval. Clin. Pr. 2012, 18, 12–18. [Google Scholar] [CrossRef]
- Thomas, B.H.; Ciliska, D.; Dobbins, M.; Micucci, S. A process for systematically reviewing the literature: Providing the research evidence for public health nursing interventions. Worldviews Evid. Based Nurs. 2004, 1, 176–184. [Google Scholar] [CrossRef]
- McMaster University School of Nursing. Quality assessment tool for quantitative studies. In Effective Public Health Practice Project (EPHPP); Assessment Tool for Quality Ratings of Research; McMaster University School of Nursing: Hamilton, ON, USA, 2011; Volume 2011. [Google Scholar]
- Jackson, N.; Waters, E. Criteria for the systematic review of health promotion and public health interventions. Health Promot. Int. 2005, 20, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Deeks, J.J.; Dinnes, J.; D’Amico, R.; Sowden, A.J.; Sakarovitch, C.; Song, F.; Petticrew, M.; Altman, D.G.; International Stroke Trial Collaborative Group; European Carotid Surgery Trial Collaborative Group. Evaluating non-randomised intervention studies. Health Technol. Assess 2003, 7, iii-173. [Google Scholar] [CrossRef] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I.; Martins, R.N.; Rumble, B.; Moir, R.; Fuller, S.; Milward, E.; Currie, J.; Ames, D.; Weidemann, A.; Fischer, P.; et al. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J. Biol. Chem. 1990, 265, 15977–15983. [Google Scholar] [CrossRef]
- Bush, A.I.; Whyte, S.; Thomas, L.D.; Williamson, T.G.; Van Tiggelen, C.J.; Currie, J.; Small, D.H.; Moir, R.D.; Li, Q.X.; Rumble, B.; et al. An abnormality of plasma amyloid protein precursor in Alzheimer’s disease. Ann. Neurol. 1992, 32, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.A.; Fine, R.E.; Johnson, R.J.; Levesque, C.A.; Rathbun, W.H.; Seetoo, K.F.; Smith, S.J.; Strohmeier, G.; Volicer, L.; Delva, L.; et al. Non-age related differences in thrombin responses by platelets from male patients with advanced Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1993, 194, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.A.; Long, H.J.; Sgro, K.; Rathbun, W.H.; McMenamin, M.E.; Seetoo, K.; Tibbles, H.; Billingslea, A.M.; Fine, R.E.; Fishman, J.B.; et al. Activated Alzheimer disease platelets retain more beta amyloid precursor protein. Neurobiol. Aging 1997, 18, 147–153. [Google Scholar] [CrossRef]
- Davies, T.A.; Long, H.J.; Tibbles, H.E.; Sgro, K.R.; Wells, J.M.; Rathbun, W.H.; Seetoo, K.F.; McMenamin, M.E.; Smith, S.J.; Feldman, R.G.; et al. Moderate and advanced Alzheimer’s patients exhibit platelet activation differences. Neurobiol. Aging 1997, 18, 155–162. [Google Scholar] [CrossRef]
- Di Luca, M.; Pastorino, L.; Cattabeni, F.; Zanardi, R.; Scarone, S.; Racagni, G.; Smeraldi, E.; Perez, J. Abnormal pattern of platelet APP isoforms in Alzheimer disease and Down syndrome. Arch. Neurol. 1996, 53, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, M.; Pastorino, L.; Bianchetti, A.; Perez, J.; Vignolo, L.A.; Lenzi, G.L.; Trabucchi, M.; Cattabeni, F.; Padovani, A. Differential level of platelet amyloid beta precursor protein isoforms: An early marker for Alzheimer disease. Arch. Neurol. 1998, 55, 1195–1200. [Google Scholar] [CrossRef]
- Padovani, A.; Pastorino, L.; Borroni, B.; Colciaghi, F.; Rozzini, L.; Monastero, R.; Perez, J.; Pettenati, C.; Mussi, M.; Parrinello, G.; et al. Amyloid precursor protein in platelets: A peripheral marker for the diagnosis of sporadic AD. Neurology 2001, 57, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Colciaghi, F.; Pastorino, L.; Archetti, S.; Corsini, P.; Cattabeni, F.; Di Luca, M.; Padovani, A. ApoE genotype influences the biological effect of donepezil on APP metabolism in Alzheimer disease: Evidence from a peripheral model. Eur. Neuropsychopharmacol. 2002, 12, 195–200. [Google Scholar] [CrossRef]
- Baskin, F.; Rosenberg, R.N.; Iyer, L.; Hynan, L.; Cullum, C.M. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology 2000, 54, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.N.; Baskin, F.; Fosmire, J.A.; Risser, R.; Adams, P.; Svetlik, D.; Honig, L.S.; Cullum, C.M.; Weiner, M.F. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch. Neurol. 1997, 54, 139–144. [Google Scholar] [CrossRef]
- Borroni, B.; Colciaghi, F.; Pastorino, L.; Pettenati, C.; Cottini, E.; Rozzini, L.; Monastero, R.; Lenzi, G.L.; Cattabeni, F.; Di Luca, M.; et al. Amyloid precursor protein in platelets of patients with Alzheimer disease: Effect of acetylcholinesterase inhibitor treatment. Arch. Neurol. 2001, 58, 442–446. [Google Scholar]
- Sanchez-Gonzalez, V.J.; Ortiz, G.G.; Gallegos-Arreola, P.; Macias-Islas, M.A.; Arias-Merino, E.D.; Loera-Castaneda, V.; Martinez-Cano, E.; Velazquez-Brizuela, I.E.; Rosales-Corral, S.A.; Curiel-Ortega, C.R.; et al. Altered beta-amyloid precursor protein isoforms in Mexican Alzheimer’s Disease patients. Dis. Markers 2006, 22, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Gu, L.; Wang, Q.; Gao, L.; Zhu, J.; Lu, X.; Zhou, F.; Zhu, D.; Zhang, H.; Xie, C.; et al. Platelet Amyloid-beta Protein Precursor (AbetaPP) Ratio and Phosphorylated Tau as Promising Indicators for Early Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 75, 664–670. [Google Scholar]
- Borroni, B.; Colciaghi, F.; Caltagirone, C.; Rozzini, L.; Broglio, L.; Cattabeni, F.; Di Luca, M.; Padovani, A. Platelet amyloid precursor protein abnormalities in mild cognitive impairment predict conversion to dementia of Alzheimer type: A 2-year follow-up study. Arch. Neurol. 2003, 60, 1740–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zainaghi, I.A.; Talib, L.L.; Diniz, B.S.; Gattaz, W.F.; Forlenza, O.V. Reduced platelet amyloid precursor protein ratio (APP ratio) predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Neural. Transm. 2012, 119, 815–819. [Google Scholar] [CrossRef]
- Padovani, A.; Borroni, B.; Colciaghi, F.; Pettenati, C.; Cottini, E.; Agosti, C.; Lenzi, G.L.; Caltagirone, C.; Trabucchi, M.; Cattabeni, F.; et al. Abnormalities in the pattern of platelet amyloid precursor protein forms in patients with mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2002, 59, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Borroni, B.; Colciaghi, F.; Lenzi, G.L.; Caimi, L.; Cattabeni, F.; Di Luca, M.; Padovani, A. High cholesterol affects platelet APP processing in controls and in AD patients. Neurobiol. Aging 2003, 24, 631–636. [Google Scholar] [CrossRef]
- Srisawat, C.; Junnu, S.; Peerapittayamongkol, C.; Futrakul, A.; Soi-ampornkul, R.; Senanarong, V.; Praditsuwan, R.; Assantachai, P.; Neungton, N. The platelet amyloid precursor protein ratio as a diagnostic marker for Alzheimer’s disease in Thai patients. J. Clin. Neurosci. 2013, 20, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Hynan, L.S.; Baskin, F.; Rosenberg, R.N. Platelet amyloid precursor protein processing: A bio-marker for Alzheimer’s disease. J. Neurol. Sci. 2006, 240, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, B.; Perani, D.; Broli, M.; Colciaghi, F.; Garibotto, V.; Paghera, B.; Agosti, C.; Giubbini, R.; Di Luca, M.; Padovani, A. Pre-clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J. Neurol. 2005, 252, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, M.; Grossi, E.; Borroni, B.; Zimmermann, M.; Marcello, E.; Colciaghi, F.; Gardoni, F.; Intraligi, M.; Padovani, A.; Buscema, M. Artificial neural networks allow the use of simultaneous measurements of Alzheimer disease markers for early detection of the disease. J. Transl. Med. 2005, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.C.; Wang, H.C.; Ko, S.Y.; Wang, P.N.; Chi, C.W.; Hong, C.J.; Lin, K.N.; Liu, T.Y. Correlation between platelet amyloid precursor protein isoform ratio and cognition in Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 77–84. [Google Scholar] [CrossRef]
- Jelic, V.; Hagman, G.; Yamamoto, N.G.; Teranishi, Y.; Nishimura, T.; Winblad, B.; Pavlov, P.F. Abnormal platelet amyloid-beta protein precursor (AbetaPP) metabolism in Alzheimer’s disease: Identification and characterization of a new AbetaPP isoform as potential biomarker. J. Alzheimers Dis. 2013, 35, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Marksteiner, J.; Humpel, C. Platelet-derived secreted amyloid-precursor protein-beta as a marker for diagnosing Alzheimer’s disease. Curr. Neurovasc. Res. 2013, 10, 297–303. [Google Scholar] [CrossRef]
- Vignini, A.; Morganti, S.; Salvolini, E.; Sartini, D.; Luzzi, S.; Fiorini, R.; Provinciali, L.; Di Primio, R.; Mazzanti, L.; Emanuelli, M. Amyloid precursor protein expression is enhanced in human platelets from subjects with Alzheimer’s disease and frontotemporal lobar degeneration: A real-time PCR study. Exp. Gerontol. 2013, 48, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.B.; Abdel-All, Z.; Dodds, S.; Andrade, J.; Alves da Silva, J.; Kalaria, R.N.; O’Brien, J.T. Platelet immunoglobulin and amyloid precursor protein as potential peripheral biomarkers for Alzheimer’s disease: Findings from a pilot study. Age Ageing 2012, 41, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.W.; Todd, S.; Craig, D.; Passmore, A.P.; Coulson, D.T.; Murphy, S.; Irvine, G.B.; Johnston, J.A. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2007, 24, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Bescos, P.; Martin-Aragon, S.; Jimenez-Aliaga, K.; Benedi, J.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Processing of the platelet amyloid precursor protein in the mild cognitive impairment (MCI). Neurochem. Res. 2013, 38, 1415–1423. [Google Scholar] [CrossRef]
- Johnston, J.A.; Liu, W.W.; Coulson, D.T.; Todd, S.; Murphy, S.; Brennan, S.; Foy, C.J.; Craig, D.; Irvine, G.B.; Passmore, A.P. Platelet beta-secretase activity is increased in Alzheimer’s disease. Neurobiol. Aging 2008, 29, 661–668. [Google Scholar] [CrossRef]
- McGuinness, B.; Fuchs, M.; Barrett, S.L.; Passmore, A.P.; Johnston, J.A. Platelet Membrane beta-Secretase Activity in Mild Cognitive Impairment and Conversion to Dementia: A Longitudinal Study. J. Alzheimers Dis. 2016, 49, 1095–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorham, P.; Bark, N.; Bjorkhem, I.; Meaney, S.; Crisby, M. Platelet alpha-and beta- secretase activities are not significantly affected by dementia or mild cognitive impairment in Swedish patients. Curr. Alzheimer Res. 2010, 7, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, A.; Decourt, B.; Walker, A.; Condjella, R.; Nural, H.; Sabbagh, M.N. Development of a specific ELISA to measure BACE1 levels in human tissues. J. Neurosci. Methods 2011, 202, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Walker, A.; Gonzales, A.; Malek-Ahmadi, M.; Liesback, C.; Davis, K.J.; Belden, C.M.; Jacobson, S.A.; Sabbagh, M.N. Can platelet BACE1 levels be used as a biomarker for Alzheimer’s disease? Proof-of-concept study. Platelets 2013, 24, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Mukaetova-Ladinska, E.B.; Abdel-All, Z.; Andrade, J.; McNally, R.J.; James, P.W.; Kalaria, R.N.; O’Brien, J.T. Increase in platelet immunoglobulin in Alzheimer’s disease is normalised following cholinesterase inhibitor treatment: Preliminary results. J. Alzheimers Dis. 2012, 32, 431–436. [Google Scholar] [CrossRef]
- Liu, H.C.; Chi, C.W.; Ko, S.Y.; Wang, H.C.; Hong, C.J.; Lin, K.N.; Wang, P.N.; Liu, T.Y. Cholinesterase inhibitor affects the amyloid precursor protein isoforms in patients with Alzheimer’s disease. Dement Geriatr. Cogn. Disord. 2005, 19, 345–348. [Google Scholar] [CrossRef]
- Sarno, T.A.; Talib, L.L.; Joaquim, H.P.; Bram, J.M.; Gattaz, W.F.; Forlenza, O.V. Protein Expression of BACE1 is Downregulated by Donepezil in Alzheimer’s Disease Platelets. J. Alzheimers Dis. 2017, 55, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Borroni, B.; Cattabeni, F.; Padovani, A.; Di Luca, M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol. Dis. 2005, 19, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Baskin, F.; Rosenberg, R.N.; Fang, X.; Hynan, L.S.; Moore, C.B.; Weiner, M.; Vega, G.L. Correlation of statin-increased platelet APP ratios and reduced blood lipids in AD patients. Neurology 2003, 60, 2006–2007. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nostrand, W.E.; Schmaier, A.H.; Farrow, J.S.; Cunningham, D.D. Protease nexin-II (amyloid beta-protein precursor): A platelet alpha-granule protein. Science 1990, 248, 745–748. [Google Scholar] [CrossRef]
- Sevush, S.; Jy, W.; Horstman, L.L.; Mao, W.W.; Kolodny, L.; Ahn, Y.S. Platelet activation in Alzheimer disease. Arch. Neurol. 1998, 55, 530–536. [Google Scholar] [CrossRef]
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine, W.B., Jr.; Michon, S.C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Eimer, W.A.; et al. Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol. Neurodegener. 2012, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Davies, T.A.; Billingslea, A.M.; Long, H.J.; Tibbles, H.; Wells, J.M.; Eisenhauer, P.B.; Smith, S.J.; Cribbs, D.H.; Fine, R.E.; Simons, E.R. Brain endothelial cell enzymes cleave platelet-retained amyloid precursor protein. J. Lab. Clin. Med. 1998, 132, 341–350. [Google Scholar] [CrossRef]
- Canobbio, I.; Visconte, C.; Oliviero, B.; Guidetti, G.; Zara, M.; Pula, G.; Torti, M. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer’s disease. Cell Signal 2016, 28, 1863–1871. [Google Scholar] [CrossRef] [Green Version]
- Weller, R.O.; Nicoll, J.A. Cerebral amyloid angiopathy: Pathogenesis and effects on the ageing and Alzheimer brain. Neurol. Res. 2003, 25, 611–616. [Google Scholar] [CrossRef]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.A.; Long, H.J.; Eisenhauer, P.B.; Hastey, R.; Cribbs, D.H.; Fine, R.E.; Simons, E.R. Beta amyloid fragments derived from activated platelets deposit in cerebrovascular endothelium: Usage of a novel blood brain barrier endothelial cell model system. Amyloid 2000, 7, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Shayo, M.; McLay, R.N.; Kastin, A.J.; Banks, W.A. The putative blood-brain barrier transporter for the beta-amyloid binding protein apolipoprotein j is saturated at physiological concentrations. Life Sci. 1997, 60, PL115–PL118. [Google Scholar] [CrossRef]
- Zhang, W.; Xiong, H.; Callaghan, D.; Liu, H.; Jones, A.; Pei, K.; Fatehi, D.; Brunette, E.; Stanimirovic, D. Blood-brain barrier transport of amyloid beta peptides in efflux pump knock-out animals evaluated by in vivo optical imaging. Fluids Barriers CNS 2013, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341, Corrigendum in 2016, 533, 278. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, M.G.; Pagni, G.; Tagliarini, C.; Marazziti, D.; Pomara, N. Platelet APP Processing: Is It a Tool to Explore the Pathophysiology of Alzheimer’s Disease? A Systematic Review. Life 2021, 11, 750. https://doi.org/10.3390/life11080750
Carbone MG, Pagni G, Tagliarini C, Marazziti D, Pomara N. Platelet APP Processing: Is It a Tool to Explore the Pathophysiology of Alzheimer’s Disease? A Systematic Review. Life. 2021; 11(8):750. https://doi.org/10.3390/life11080750
Chicago/Turabian StyleCarbone, Manuel Glauco, Giovanni Pagni, Claudia Tagliarini, Donatella Marazziti, and Nunzio Pomara. 2021. "Platelet APP Processing: Is It a Tool to Explore the Pathophysiology of Alzheimer’s Disease? A Systematic Review" Life 11, no. 8: 750. https://doi.org/10.3390/life11080750
APA StyleCarbone, M. G., Pagni, G., Tagliarini, C., Marazziti, D., & Pomara, N. (2021). Platelet APP Processing: Is It a Tool to Explore the Pathophysiology of Alzheimer’s Disease? A Systematic Review. Life, 11(8), 750. https://doi.org/10.3390/life11080750