
Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation

 and
and 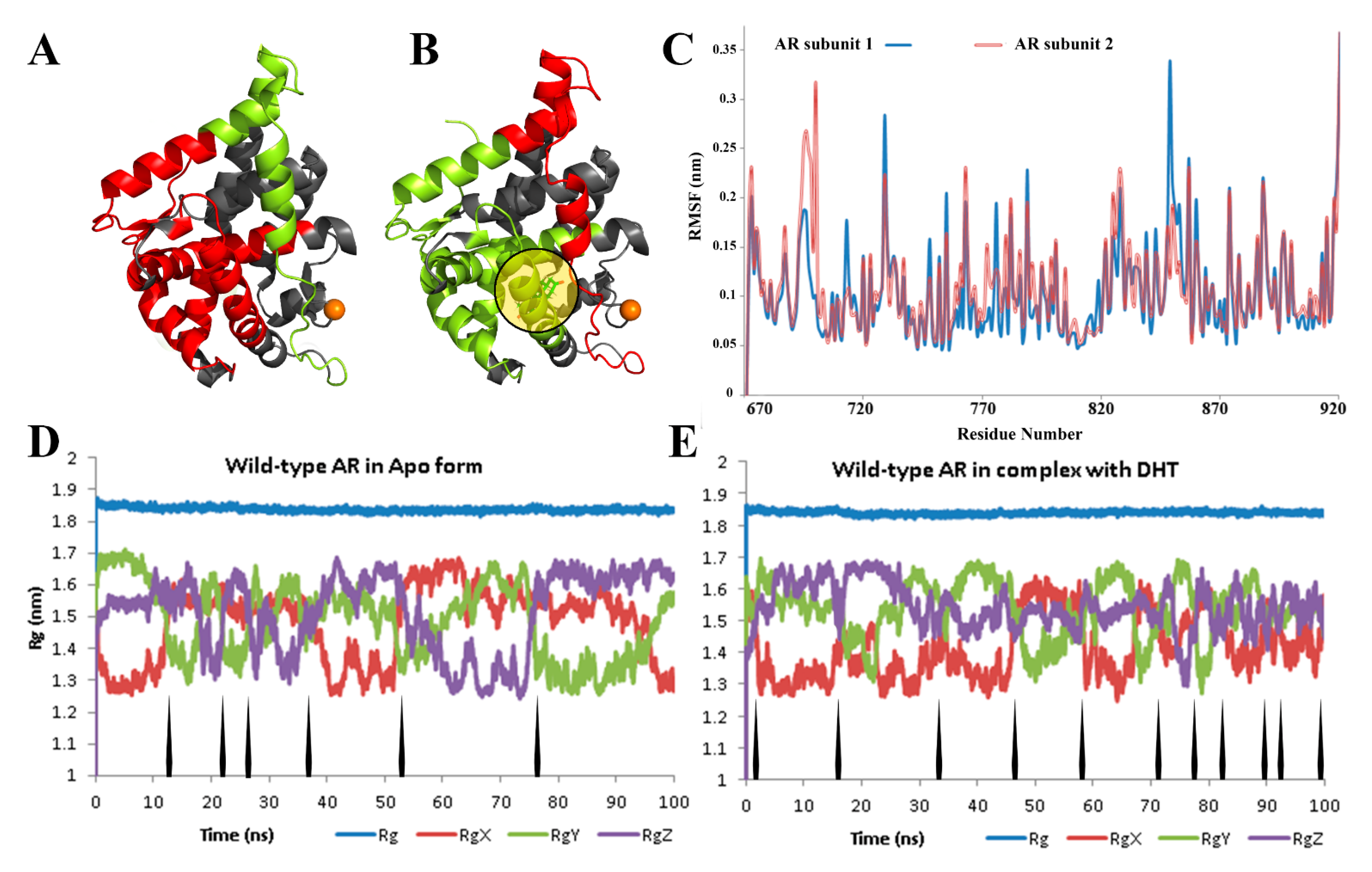{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
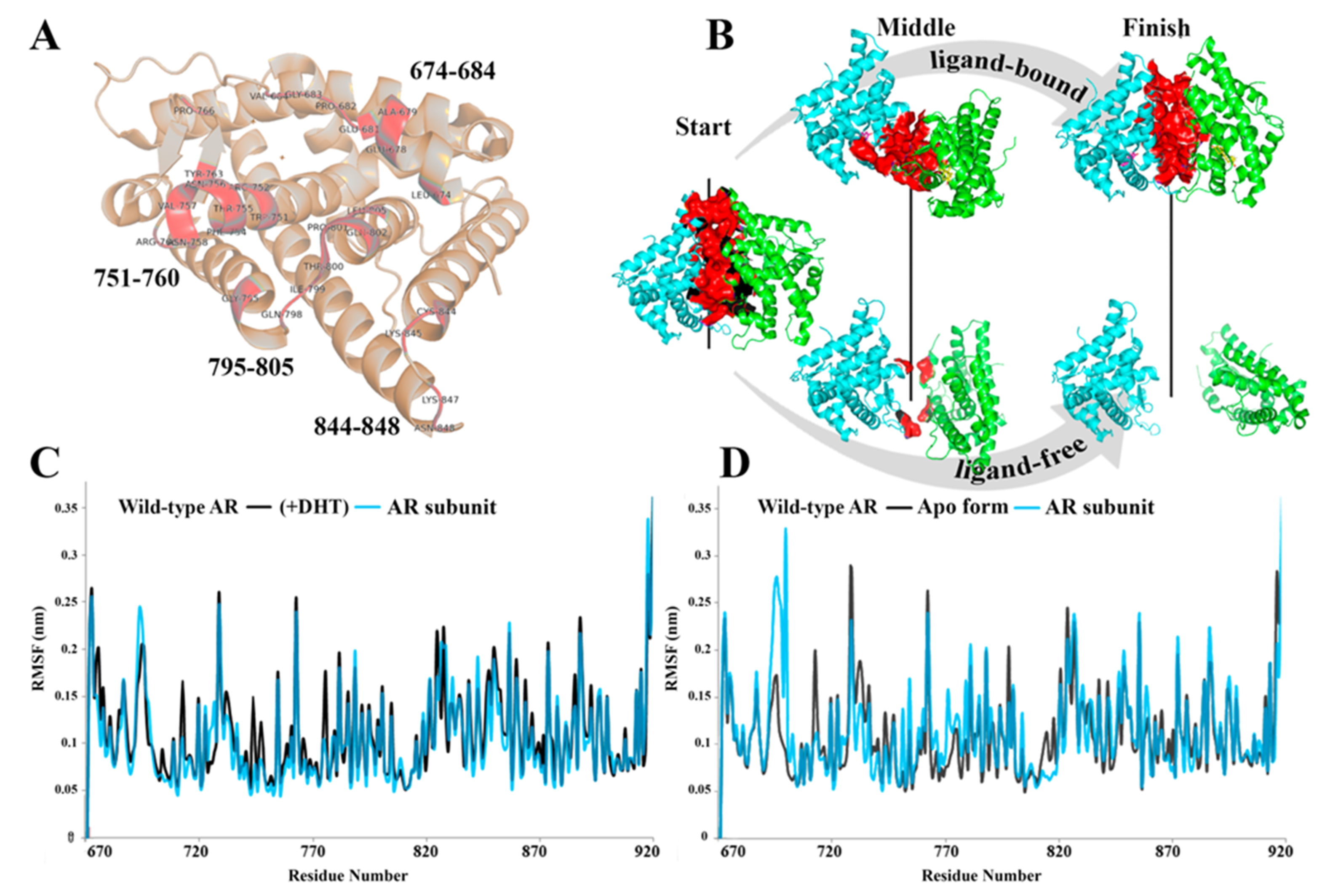
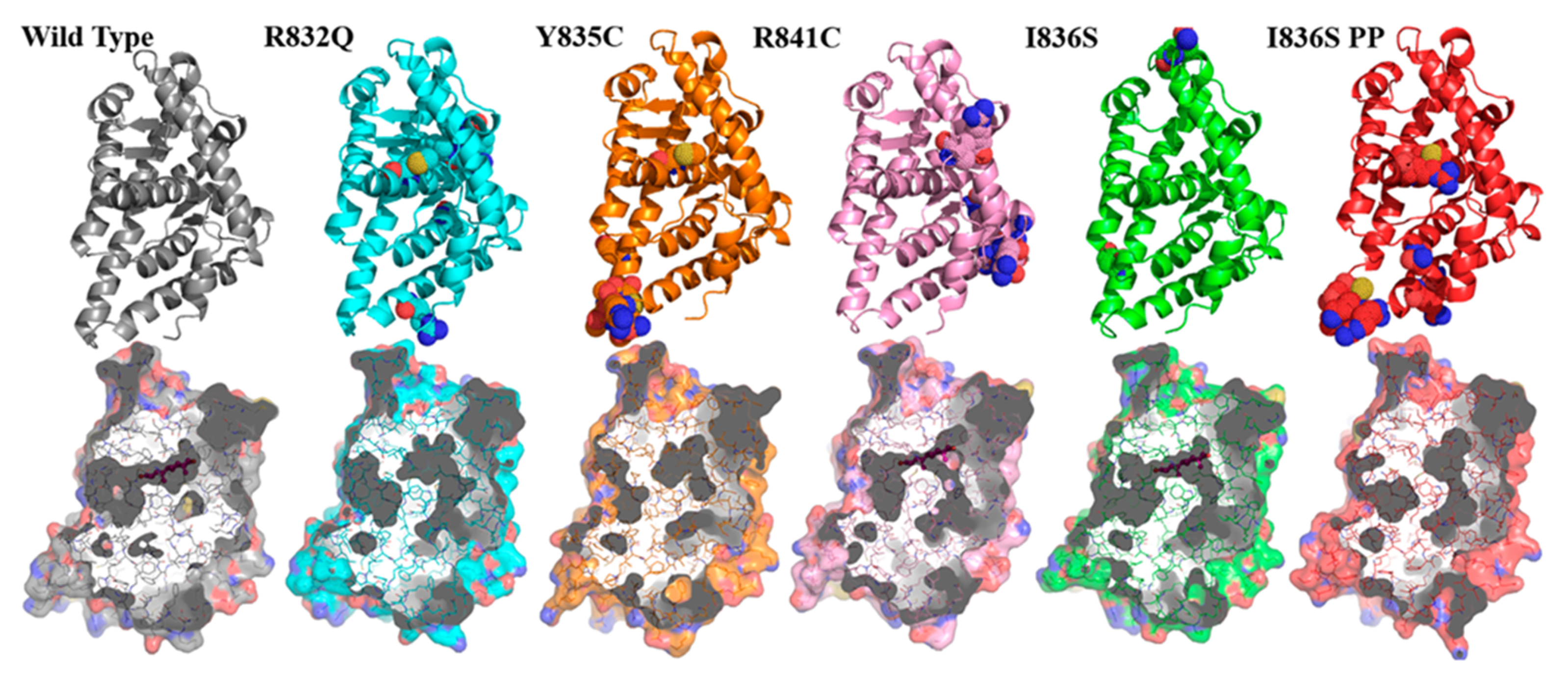
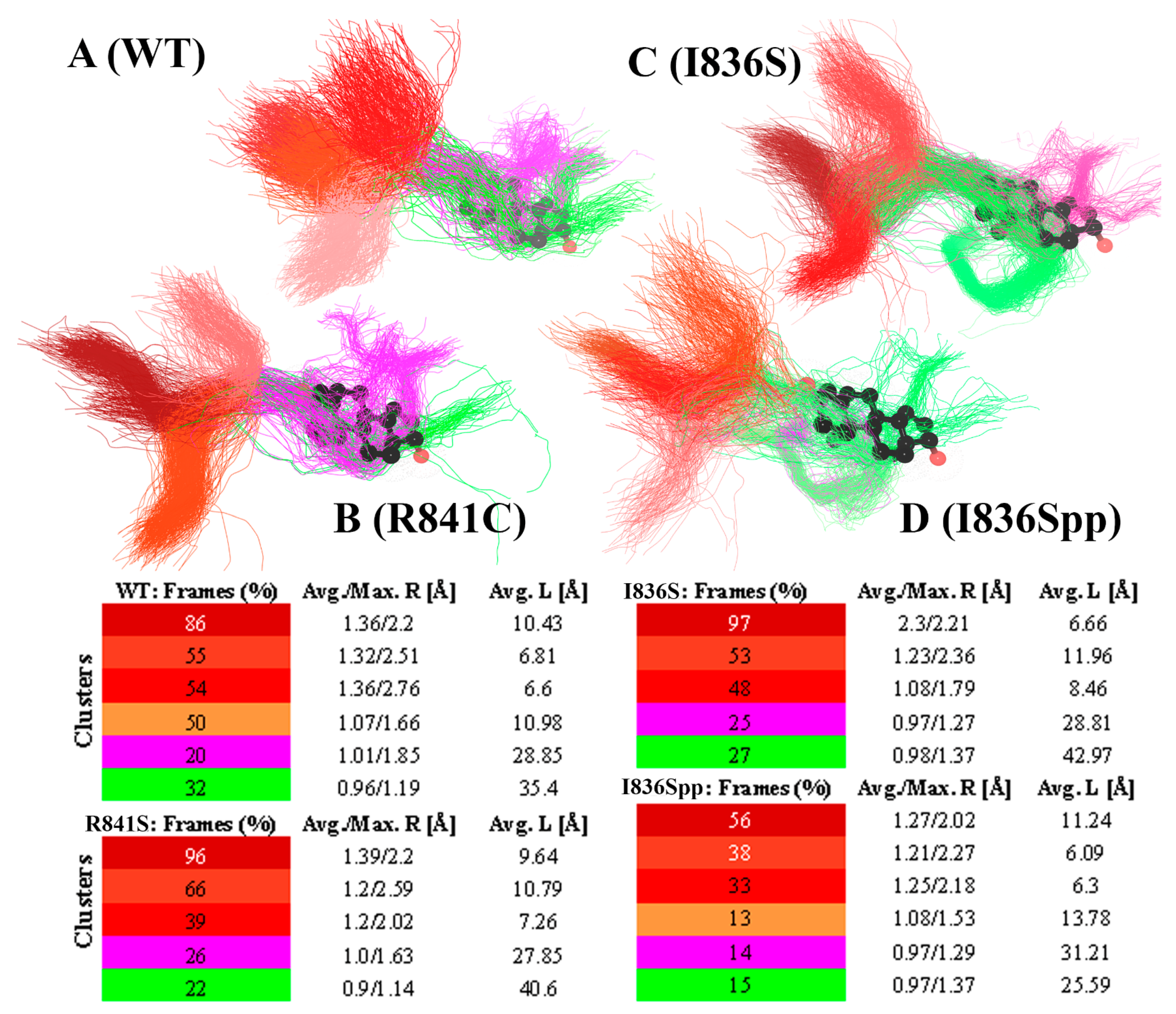
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oakes, M.B.; Eyvazzadeh, A.D.; Quint, E.; Smith, Y.R. Complete androgen insensitivity syndrome—A review. J. Pediatr. Adolesc. Gynecol. 2008, 21, 305–310. [Google Scholar] [CrossRef]
- Galani, A.; Kitsiou-Tzeli, S.; Sofokleous, C.; Kanavakis, E.; Kalpini-Mavrou, A. Androgen insensitivity syndrome: Clinical features and molecular defects. Hormones (Athens) 2008, 7, 217–229. [Google Scholar] [CrossRef]
- Bevan, C.L.; Hoare, S.; Claessens, F.; Heery, D.M.; Parker, M.G. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell Biol. 1999, 19, 8383–8392. [Google Scholar] [CrossRef] [PubMed]
- Berrevoets, C.A.; Doesburg, P.; Steketee, K.; Trapman, J.; Brinkmann, A.O. Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2). Mol. Endocrinol. 1998, 12, 1172–1183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ikonen, T.; Palvimo, J.J.; Jänne, O.A. Heterodimerization is mainly responsible for the dominant negative activity of amino-terminally truncated rat androgen receptor forms. FEBS Lett. 1998, 430, 393–396. [Google Scholar] [CrossRef]
- Karvonen, U.; Kallio, P.J.; Jänne, O.A.; Palvimo, J.J. Interaction of androgen receptors with androgen response element in intact cells. Roles of amino- and carboxyl-terminal regions and the ligand. J. Biol. Chem. 1997, 272, 15973–15979. [Google Scholar] [CrossRef]
- Langley, E.; Kemppainen, J.A.; Wilson, E.M. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J. Biol. Chem. 1998, 273, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Shizu, R.; Yokobori, K.; Perera, L.; Pedersen, L.; Negishi, M. Ligand induced dissociation of the AR homodimer precedes AR monomer translocation to the nucleus. Sci. Rep. 2019, 9, 16734. [Google Scholar] [CrossRef]
- Meehan, K.L.; Sadar, M.D. Androgens and androgen receptor in prostate and ovarian malignancies. Front. Biosci. 2003, 8, d780–d800. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef]
- Gioeli, D.; Paschal, B.M. Post-translational modification of the androgen receptor. Mol. Cell Endocrinol. 2012, 352, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Myers, M.; Brown, M. Formation of the androgen receptor transcription complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Sirokha, D.; Gorodna, O.; Lozhko, D.; Livshyts, G.; Zelinska, N.; Livshits, L. Novel missense mutation in ligand binding domain of AR gene identified in patient with androgen insensitivity syndrome from Ukraine. Clin. Case Rep. 2021, 9, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nature Methods 2014, 11, 603–604. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Patrick, R.; Kobe, B.; Lê Cao, K.-A.; Bodén, M. PhosphoPICK-SNP: Quantifying the effect of amino acid variants on protein phosphorylation. Bioinformatics 2017, 33, 1773–1781. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- García, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef]
- Falsafi-Zadeh, S.; Karimi, Z.; Galehdari, H. VMD DisRg: New User-Friendly Implement for calculation distance and radius of gyration in VMD program. Bioinformation 2012, 8, 341–343. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef]
- Pavelka, A.; Sebestova, E.; Kozlikova, B.; Brezovsky, J.; Sochor, J.; Damborsky, J. CAVER: Algorithms for Analyzing Dynamics of Tunnels in Macromolecules. IEEE/ACM Trans. Comput Biol. Bioinform. 2016, 13, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Grino, P.B.; Griffin, J.E.; Wilson, J.D. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 1990, 126, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Guiochon-Mantel, A.; Delabre, K.; Lescop, P.; Milgrom, E. The Ernst Schering Poster Award. Intracellular traffic of steroid hormone receptors. J. Steroid Biochem. Mol. Biol. 1996, 56, 3–9. [Google Scholar] [CrossRef]
- Prescott, J.; Coetzee, G.A. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 2006, 231, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Schaufele, F.; Carbonell, X.; Guerbadot, M.; Borngraeber, S.; Chapman, M.S.; Ma, A.A.K.; Miner, J.N.; Diamond, M.I. The structural basis of androgen receptor activation: Intramolecular and intermolecular amino-carboxy interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 9802–9807. [Google Scholar] [CrossRef] [PubMed]
- Anne Etgen, D.W.P. Molecular Mechanisms of Hormone Actions on Behavior, 1st ed.; Academic Press: Cambridge, MA, USA, 2009; p. 1100. [Google Scholar]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Lane, M.V.; Kemppainen, J.A.; French, F.S.; Wilson, E.M. Specificity of ligand-dependent androgen receptor stabilization: Receptor domain interactions influence ligand dissociation and receptor stability. Mol. Endocrinol. 1995, 9, 208–218. [Google Scholar] [CrossRef][Green Version]
- Pereira De Jésus-Tran, K.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef] [PubMed]
- Lusher, S.J.; Raaijmakers, H.C.A.; Vu-Pham, D.; Kazemier, B.; Bosch, R.; McGuire, R.; Azevedo, R.; Hamersma, H.; Dechering, K.; Oubrie, A.; et al. X-ray structures of progesterone receptor ligand binding domain in its agonist state reveal differing mechanisms for mixed profiles of 11β-substituted steroids. J. Biol. Chem. 2012, 287, 20333–20343. [Google Scholar] [CrossRef]
- Williams, S.P.; Sigler, P.B. Atomic structure of progesterone complexed with its receptor. Nature 1998, 393, 392–396. [Google Scholar] [CrossRef]
- Su, L.; Cheng, J.; Yin, X.; Liu, G.; Lu, Z.; Sheng, H.; Cai, Y.; Shi, Q.; Liu, L. Clinical and molecular characteristics in 15 patients with androgen receptor gene mutations from South China. Andrologia 2017, 49. [Google Scholar] [CrossRef]
- Yaegashi, N.; Uehara, S.; Senoo, M.; Sato, J.; Fujiwara, J.; Funato, T.; Sasaki, T.; Yajima, A. Point mutations in the steroid-binding domain of the androgen receptor gene of five Japanese patients with androgen insensitivity syndrome. Tohoku J. Exp. Med. 1999, 187, 263–272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilson, C.M.; Griffin, J.E.; Wilson, J.D.; Marcelli, M.; Zoppi, S.; McPhaul, M.J. Immunoreactive androgen receptor expression in subjects with androgen resistance. J. Clin. Endocrinol. Metab. 1992, 75, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, J.; Yong, E.L. Ligand- and coactivator-mediated transactivation function (AF2) of the androgen receptor ligand-binding domain is inhibited by the cognate hinge region. J. Biol. Chem. 2001, 276, 7493–7499. [Google Scholar] [CrossRef] [PubMed]
- Duff, J.; McEwan, I.J. Mutation of Histidine 874 in the Androgen Receptor Ligand-Binding Domain Leads to Promiscuous Ligand Activation and Altered p160 Coactivator Interactions. Mol. Endocrinol. 2005, 19, 2943–2954. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayevsky, A.; Sirokha, D.; Samofalova, D.; Lozhko, D.; Gorodna, O.; Prokopenko, I.; Livshits, L. Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation. Life 2021, 11, 659. https://doi.org/10.3390/life11070659
Rayevsky A, Sirokha D, Samofalova D, Lozhko D, Gorodna O, Prokopenko I, Livshits L. Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation. Life. 2021; 11(7):659. https://doi.org/10.3390/life11070659
Chicago/Turabian StyleRayevsky, Alexey, Dmytro Sirokha, Dariia Samofalova, Dmytro Lozhko, Olexandra Gorodna, Inga Prokopenko, and Liudmyla Livshits. 2021. "Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation" Life 11, no. 7: 659. https://doi.org/10.3390/life11070659
APA StyleRayevsky, A., Sirokha, D., Samofalova, D., Lozhko, D., Gorodna, O., Prokopenko, I., & Livshits, L. (2021). Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation. Life, 11(7), 659. https://doi.org/10.3390/life11070659