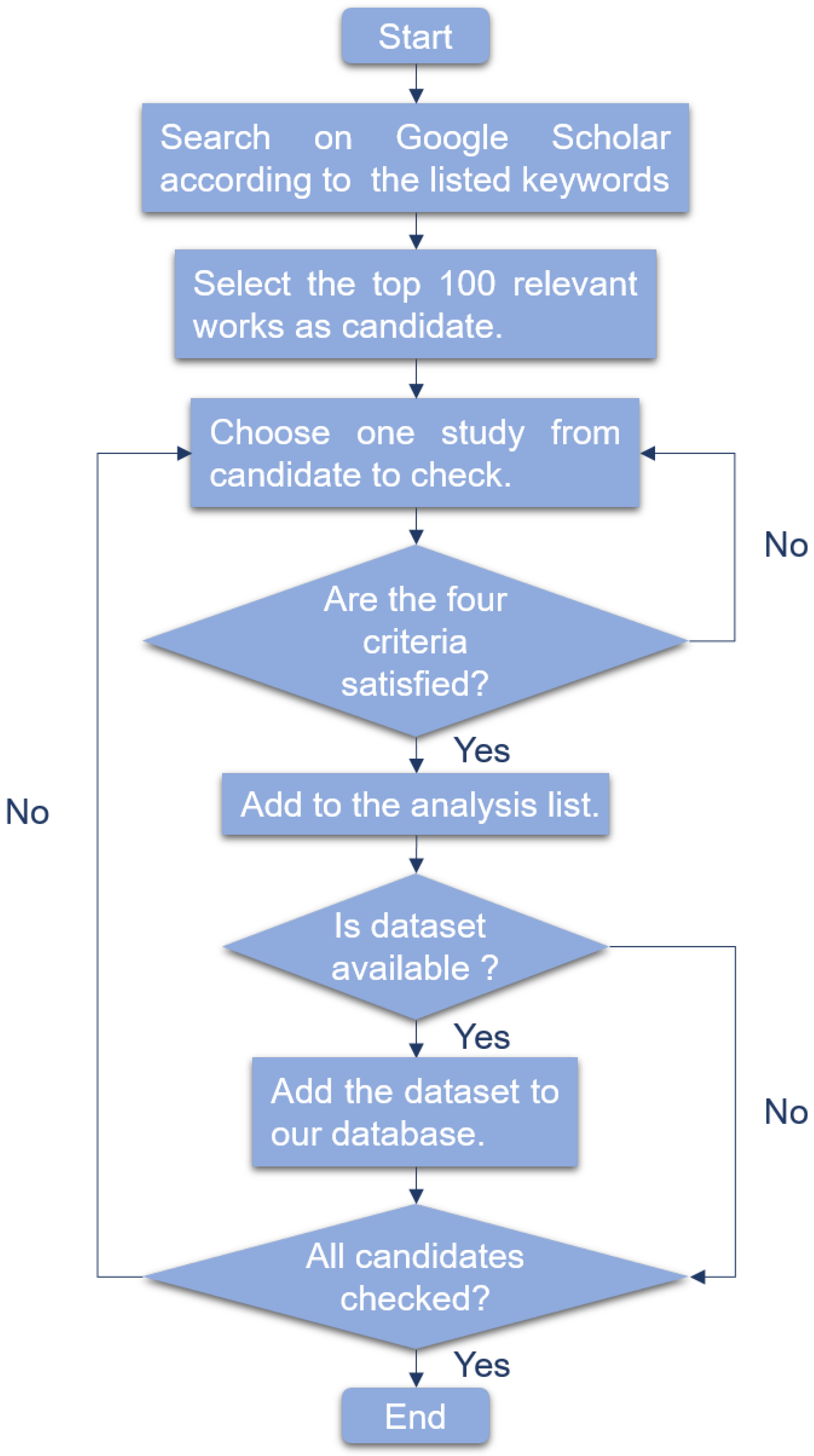
For traditional machine learning algorithms, we reviewed linear models, support vector machine and random forest.
4.1.1. Linear Models
Linear models are widely used for supervised learning because of the advantage of implementation simplicity and interpretability. Linear regression, logistic regression and LASSO are some examples of linear models.
Given an input data for . Let denote the prediction made by a model for the given input. Coefficients () are parameters that define the model by assigning a coefficient to each input, and the bias or intercept is provided by an additional coefficient. The training data is used to estimate the coefficients of the logistic regression algorithm using a learning algorithm known as a maximum-likelihood estimation. The learning algorithm assumes data distribution and produces coefficients that minimize the error of probabilities of model prediction to those in the data.
The logistic regression model can be described with a matrix for the input data
X, a vector for the output
, and a vector for the coefficients
using linear algebra represented as the Formula (
33).
Since the above representation is identical to linear regression, which produces real values as outputs instead of class labels, a nonlinear function is used to ensure that the output of the weighted sum is a value between 0 and 1.
Logistic regression uses the logistic function, also known as the sigmoid function, to ensure class labels’ prediction. The sigmoid function is an S-shaped curve that maps a real-valued number
x into a number between 0 and 1 using Equation (
34).
Therefore, for logistic regression,
x in Equation (
34) is replaced with the weighted sum given in Equation (
35) to produce an output between 0 and 1 for two class labels 0 and 1.
The output from the model can be interpreted as a probability from a Binomial probability distribution function.
Least Absolute Shrinkage and Selection Operator (LASSO), also known as L1-norm, adds a regularization term which is used to penalize the less important features in a data by making their respective coefficient (
) zero, thereby shrinking their weights to zero. The less important features in Equation (
33) having
are eliminated, thereby making LASSO useful for feature selection and the creation of simple models. It is beneficial for datasets with high dimensions and high correlation. L1-norm is given by Equation (
36)
where
is the hyperparameter that controls the shrinkage. The bias of the model increases as
increases while variance increases as
decreases.
- B.
The Application of Linear Models in Early Cancer Detection
Linear models have been applied in many ways to detect several types of cancer, either recurrent or metastatic, in different parts of the body.
Table 6 is an overview of relevant publications based on linear models.
Maltoni et al. [
150] used a logistic regression model to evaluate the role of altered genes in breast cancer like HER2, PI3KCA for patient prognosis due to the possibility of their correlation with CF-DNA quantity. They collected serum samples from 58 non-relapsed and 21 relapsed patients and analyzed the samples for cfDNA integrity and quantity of all oncogenes. To determine the ability of these genes in predicting a relapse, the logistic regression on a two-marker combination produced an area under curve of 0.627 with a 95% confidence interval. With further clinical validity, the study speculates the potential of cfDNA detected as liquid biopsy in clinical practice.
Gene expression information of original tissues is contained in the nucleosome footprint of cfDNA. This information can be used in the prediction of response to chemotherapy. Yang et al. [
149] utilized LASSO to evaluate transcription start site (TSS) regions coverage ability of genes. Based on cfDNA data of 85 healthy individuals and 85 individuals who are breast cancer patients, the coverage at the TSS regions was utilized for the classification of individuals into either having cancer or healthy. The LASSO model was repeated 100 times with a 5-fold cross-validation technique using the R package to prevent bias. A test was implemented using plasma from 30 healthy donors and 60 patients to validate the model independently. The model recorded a significant median AUC of 0.863 for the training cohort and 0.834 for the validation cohort. The model was able to avoid overfitting, as noticed in the recorded AUC. With the analysis, the use of cfDNA nucleosome footprints to predict neoadjuvant chemotherapy was highlighted and verified with the LASSO model. The study will improve personalized decision-making per patients’ treatment.
Due to the advancement of lung cancer by the time it is diagnosed, it is the deadliest cancer in the world [
151]. El-Khoury et al. [
148] used the bootstrap sampling method and LASSO penalization to deduce the suitable combination of protein necessary for predicting outcome to improve early detection and patients’ survival. With data comprising 93 healthy donors and 128 lung cancer patients, the level of plasma in 351 proteins was quantified, and the optimal threshold for the biomarker was selected. The validation of the panel was carried out with independent data of 49 healthy donors and 48 patients using logistic regression. With an AUC of 0.999, sensitivity of 0.992, specificity of 0.989, negative predictive value of 0.989 and positive predictive value of 0.992, lung cancer was detected irrespective of the cancer stage, making it possible to detect lung cancer earlier and aiding early treatment.
For early and accurate decisions on treatment strategies, an accurate diagnosis must be made. Therefore, it is vital to distinguish small cell lung cancer (SCLC) from non-small cell lung cancer (NSCLC). Non-small cell lung cancer can be further categorized as squamous cell carcinoma and inter alia adenocarcinoma. Raman et al. [
147] collected public data containing 843 samples (small cell lung cancer = 68, squamous cell carcinoma = 351, and inter alia adenocarcinoma = 424) which were filtered based on histology. cfDNA was extracted was further extracted from plasma. Five classifiers, including random forest, support vector machine, multinomial logistic regression with ridge regularization, multinomial logistic regression with elastic net regularization, and multinomial logistic regression with lasso regularization were evaluated with the data using a leave-one-out cross-validation method. Due to the inability of some classifiers to deal with class imbalance, the authors used a random sampling method to make the number of samples in all classes equal to 68 to make the number of training samples equal to 204. Multinomial logistic regression with ridge regularization, based on iterative one-vs.-all receiver operating curve, had the best performance with a mean area under curve of 0.936. The coefficients of the logistic regression model detected that the prominent regions which differentiate non-small lung cell cancer from small lung cell cancer are located at the chromosome arm, and tumor fraction is a determinant of the prediction probability.
Cucchiara et al. [
145], working with the metastatic case of EGFR-positive NSCLC reports the possibility of using the combination of liquid biopsy and radiomics to suggest management of the disease. This can be done by detecting new mutations early. Liquid biopsy is easy to perform, minimally invasive and can be done repeatedly to extract valuable information. cfDNA acquired from plasma of seven metastatic patients was analyzed using digital droplet PCR, and radiomic analysis was also done using computed tomography images. The authors were able to compare the EGFR mutation dynamics in cfDNA with the radiomic features. They used a logistic LASSO regression model to estimate the correlation between the variation in the radiomics features and the EGFR mutation status using a 27-fold Monte Carlo cross-validation method. The model implemented a feature reduction, and maximum likelihood estimation was done for the remaining features. Based on these performance analyses, an early decision can be made for treatment strategy. Although the authors found no significant relationship between the mutational status and tumor volume, there was also no discovered association between the clinical outcomes and the radiomic signatures.
Wei et al. [
146] pointed out the need to have less invasive strategies for the early prognosis and detection of colorectal cancer to avoid distant metastasis. The authors extracted extracellular vesicles from plasma samples and used nanoparticle tracking analysis, transmission electron microscopy with western blotting to identify the extracellular vesicles. The samples contained 37 colorectal cancer patients, 22 colorectal adenoma patients and 42 non-cancerous control participants. It was discovered that circulating EV-miR-193a-5p can efficiently distinguish the three classes. Especially with an AUC of 0.752, it can distinguish colorectal cancer patients from the two other classes and with an AUC of 0.759, it can distinguish colorectal cancer from non-cancer. This shows circulating EV-miR-193a-5p can identify colorectal cancer than precancerous lesions. In addition, due to the importance of age factor in colorectal cancer, a logistic regression model was implemented to integrate the age with a cutoff of 55 years and circulating EV-miR-193a-5p. The integration of the age factor increased the area under curve from 0.752 to 0.775 and 0.759 to 0.795 for distinguishing colorectal cancer patients from the two other classes and colorectal cancer from non-cancer, respectively. The integration of the age factor using the model can quickly identify colorectal cancer in high-risk individuals.
Oral cancer, being one of the most frequent cancer in the world, Lin et al. [
143] identified the correlation between the progression of oral squamous cell carcinoma and cfDNA. The identification of the biomarkers is essential to improve diagnosis and treatment. Plasma was extracted from 121 oral cancer patients and 50 individuals for control while ensuring that the cfDNA size distribution is similar in oral cancer patients and control donors. Analyses on the dataset revealed that the mean concentration of cfDNA in oral cancer patients was significantly higher than that of the control group. The adjusted odds ratios were determined using binary logistic regression analysis, and a confidence interval of 95% was achieved. With a statistical significance test of
p < 0.05, the study established the relationship between cfDNA and oral cancer.
Due to the role that serum exosome plays in the development of cancer, Li et al. [
144] identified protein content in serum exosome based on 30 samples. The samples included oral cancer patients with lymph node metastasis, oral cancer patients with no lymph node metastasis, and healthy controls. Oral cancer patients have a high rate of lymph node metastasis [
152]. A binary logistic regression analysis was carried out to compare the use of four biomarkers (ApoA1, CXCL7, PF4V1, F13A1) and their combinations based on the area under curve. This study deduced that the four biomarkers from serum exosomes could help diagnose oral cancer-lymph node metastasis.
Due to the lack of early detection and resistance to chemotherapy, ovarian cancer is the most lethal cancer in gynecology [
153,
154]. Li et al. [
142] performed a two-stage epigenome-wide association study to identify methylation biomarkers for epithelial ovarian cancer. The authors selected 24 cancer cases, and 24 age-frequency matched control cases for genome-wide methylation profiling, and 206 cancer cases with 205 age-frequency matched control cases. Independent
t-test and
test was used for the continuous and categorical variables, respectively. The correlation between the blood cell counts and the DNA methylation was estimated using Pearson correlation analysis. A logistic regression model was further built for the differentially methylated cpG sites in the validation stage, and it was evaluated based on the receiver operating characteristics curves. With the study, the identified set of blood-derived DNA methylation signatures and its association with epithelial ovarian cancer will serve as a tool for the early detection of ovarian cancer.
Linear models have been successfully applied to different cancer types, including breast cancer, colorectal cancer, oral cancer, lung cancer, etc. Ranging from classification to the selection of important features for further prognosis, the application of linear models as machine learning tools is important.
4.1.2. Support Vector Machine
Support Vector Machine (SVM) [
155] is a supervised learning method for solving data mining problems, first proposed by Cortes and Vapnik in 1995. It aims to build a decision boundary, which is known as the hyperplane, to separate different classes. The positive samples and negative samples each have the closest point to the hyperplane. SVM distinguishes different classes by maximizing the distance between these two points to the hyperplane.
If the data instances are
for
, where
and
. The two classes in the training data can be separated by a hyperplane
H:
. Furthermore, there are two hyperplanes
:
and
:
parallel to
H. The positive and negative samples, which are closest to
H, just fall on
and
, respectively. Such samples are support vectors. Margin is defined as the distance between
and
in Formula (
37).
SVM aims to learn an optimal separating hyperplane
H to maximize the margin (minimize
), while keeping all the points correctly classified. This problem can be summarized as Formula (
38).
For non-separable data, slack variable
is defined to allow data samples to violate the margin or even misclassified. In Formula (
39),
C is the penalty parameter.
When the true model of the dataset is nonlinear, we can map the input data
into a new high dimensional space
employing a nonlinear mapping
. After mapping, the problem can be summarized as Formula (
40).
To solve this problem, we need to rewrite the primal problem into its dual form.
In Formula (
41),
is the Lagrange Multiplier. The SVM dual problem contains the inner product of
, which is the high-dimensional feature vector. To simplify the calculation, kernel function is defined to replace the inner product as Formula (
42).
- B.
The Application of SVM in Early Cancer Detection
As a traditional and popular machine learning method, SVM was widely used for early cancer detection. An overview of relevant reference to SVM is provided in
Table 7.
Patrick et al. [
156] reported a work of glioblastoma detection utilizing SVM with radial basis kernel. In this study, 1158 miRNAs collected from blood were analyzed. They applied SVM and filter based feature selection method to determine a suitable subset of miRNA biomarkers and achieved their best result based on 180 miRNAs with an accuracy of 81%, specificity of 79%, and sensitivity of 83%. Additionally, 52 miRNAs were significantly distinguished by unpaired Student’s
t-test. On this basis, miR-128 and miR-342-3p stand out significantly with a
p-value of 0.025 under correcting for multiple testing by Benjamini-Hochberg adjustment. This work revealed the possibility of miR-128, miR-342-3p and other important miRNA as biomarkers to detect glioblastom based on the analyses of 20 patients and 20 healthy individuals. It is also an instance of the effectiveness of SVM on a small sample dataset with high dimensions.
In 2015, Thomas Wurdinger’s team from the Netherlands published a study in Cancer Cell showing that mRNA from tumor-educated platelets (TEPS) is potential for diagnosis of various cancers and differentiation of cancer types [
141]. This is the first time that the term of tumor-educated platelet proposed. They identified that 1453 mRNAs increased and 793 mRNAs decreased in TEPs compared with healthy platelets. Further analysis indicated that the increased TEP mRNAs were involved in biological processes such as vesicle-mediated transport and the cytoskeletal protein binding while the decreased mRNAs were involved in RNA processing and splicing. A pan-cancer classification based on SVM was implemented, distinguishing 228 patients in 6 cancers from 55 healthy individuals with 96% accuracy. Additionally, TEP mRNA profiles are also demonstrated to be effective in distinguishing the specific tumor type. Besides, they found that the platelet samples of patients possess distinct therapy-guiding markers confirmed in matching tumor tissue. In their further study [
157], this team combined particle-swarm optimization (PSO) and SVM to detect non-small-cell lung cancer based on TEPs. PSO was utilized to identify the optimal biomarker panels from large amounts liquid biosources and to tune the parameter of SVM. They termed this pipeline PSO-enhanced thromboSeq. In 2019, they reevaluated the publicly available dataset in [
157] and further validate the performance on a new platelet-RNA-sequencing dataset from a healthy donor (HD) and lower-grade glioma (LGG) samples [
159]. In this manuscript, the authors not only provided a new dataset but also disclosed the code and state the operation of the code step by step. Heinhuis et al. [
163] generalized the pipeline of PSO-enhanced thromboSeq to identify the biomarker for sarcoma on a dataset with 160 samples, achieving a diagnostic accuracy of 87% and AUC of 0.93.
Cario et al. [
166] diagnosed oral cancer based on the Fourier-transform infrared (FTIR) spectra of salivary exosomes. The dataset is the whole saliva samples collected from 21 oral cancer patients and 13 healthy individuals. By analyzing the absorbance spectra, they found a number of differences between normal and cancer samples, including changes in the conformations of proteins, lipids and nucleic acids. Based on these findings, this work adopted the spectra absorbance bands between the 900 cm
and 3700 cm
, the ratios and the area under the absorbance spectrum of different three certain band as the input features of classifiers. Principal component analysis–linear discriminant analysis (PCA–LDA) and SVM are included as the discrimination models. In terms of accuracy, SVM achieved a training accuracy of 100% and a cross-validation accuracy of 89%. PCA–LDA showed an accuracy of 95%.
Sunkara et al. [
160] presented a centrifugal device for isolation of extracellular vesicles (EVs) from whole-blood. SVM was utilized to analyze the 8 biomarkers to detect 43 prostate-cancer patients from 30 healthy individuals. HSP90 achieved the highest sensitivity (86%), accuracy (88%), specificity (90%), and AUC (0.92) of all the test markers.
Guangzhe et al. [
161] applied SVM to detect urothelial carcinoma (UC) from 65 patients with urothelial carcinoma, 58 with kidney cancer, 45 with prostate cancer, and 95 normal individuals by analyzing copy number alterations of urinary cfDNA.In this work, the random forest was first utilized to select the top 50 features. After feature selection, RF, SVM and LASSO were compared and SVM with linear kernel outperformed the other two models. The authors defined UCdetector as a combination of the 50 CNA features selected by the RF and the SVM classifier with linear kernel. UCdetector achieved the AUC of 0.959 under 10 repeats of random splitting on this dataset. Further validation on an independent dataset comprising 24 normal samples and 28 UC patients was implemented. The UCdetector distinguishes UC with an AUC of 0.888. To test the clinical sensitivity of selected 50 CNA features, the authors applied UCdetector on the 410 patients from TCGA and 90 patients from Chinese UTUC WGS data. UCdetector could accurately identify the upper tract urothelial cancers at the AUC of 0.996. Furthermore, the concordance performance of urinary cfDNA was reported to be more sensitive than the urinary sediment. This recent work recognized the top 50 important CNA features from 5000 original features and achieved satisfying performance on different datasets, even on tissue samples from TCGA. For further comment, it demonstrates the power of feature selection based on RF and the identity capacity of SVM.
Shicai et al. [
162] combined SALP-seq and SVM as a pipeline to discover new cfDNA-based biomarkers for esophageal cancer. They studied the reads density of all promoters and found high reads density in normal samples and extremely low-density cancer samples on 49 genes. Of these, 34 genes are newly discovered biomarkers. The author further validated the relationship between esophageal cancer and these biomarkers on a dataset with 163 esophageal cancer samples and 11 normal samples. Moreover, 88 important regions associated with esophageal cancer were screened out from the whole genome and 54 of these, located in distal intergenic and proximal regulatory regions, were inferred to be potential diagnostic and prognostic markers for cancer. Additionally, 37 mutated genes, unique in pre-operation patients, were also discovered from a large amount of mutations in thousands of genes in pre- and post-operated esophageal cancer samples and normal samples. In this work, 103 epigenetic markers and 37 genetic markers were discovered for esophageal cancer. Finally, SVM was adopted to detect cancer samples based on 88 cancer-associated regions and achieved a high AUC of 1.0.
Zhang et al. [
164] designed a DNA molecular computation platform involving SVM to analyze miRNA profiles from serum samples. They validate the performance based on clinical serum samples from 8 healthy individuals and 14 lung cancer patients with an accuracy of 86.4%.
In our recently published work [
165], we proposed an Adaptive Support Vector Machine (ASVM) method by combining Shuffled Frog Leaping Algorithm and SVM for pan-cancer and subsequent tumor origin analysis. The proposed method was firstly validated on a cell-free DNA dataset with 423 sample records. We observed an improvement of AUC from 0.832 for SVM to 0.938 for ASVM. The proposed ASVM was competitive or outperformed the other six machine learning models on both the original dataset and additional two datasets.
4.1.3. Random Forest
Random Forest (RF) is an ensemble machine learning approach consisting of randomly selected decision tree subsets for classification and regression. Leo Breiman introduced a random forest algorithm using bootstrapping in the random tree selection method in the early 2000s [
167]. It was an enormous improvement in classification and regression machine learning accuracy. It uses the bag of random tree classifications to the ensemble and evaluates the overall classification for the given training and test data set.
The basic principle of the RF algorithm is the bootstrapping aggregation of randomly selected decision trees from given data observations. According to the Breiman [
167] RF algorithm, it deals with classification and regression tasks using the random forest to learn. For the general RF regression estimation, Let
X is the random input vector, where
. We need to predict the response
Y using the following Equation (
43).
Now training sample
for independent input and goal data pairs of
dataset that construct estimate with random tree
T for
m Function (
43). Now RF consists of
M numbers of random regression trees. The predicted estimation value (
) for the
tree input
x is defined as:
where
is the set of input data points for each tree and
is the data elements of each input observation,
is preselected data for input tree construction from
. Now final random forest estimation is:
For RF supervised classification, it can classify both binary and multi-class datasets [
168]. Let input vector and Y is a random class vector with class value 0,1. Now we can predict label Y from input X and
dataset. Therefore, RF binary classifier can obtain from the random classification trees as:
- B.
The application of Random Forest in Early Cancer Detection
In recent years, several studies employed RF for early cancer detection from different liquid biopsy data. An overview of relevant references is provided in
Table 8.
Song CX et al. [
169] applied the RF algorithm to predict lung cancer, pancreatic cancer, and HCC using cfDNA 5-Hydroxy-methyl-cytosine (5hmC) mark in blood plasmas. This study collected the whole-genome cfDNA 5hmC signatures from 49 patients with seven cancer types and eight healthy individuals for their sequence analysis using the 5hmC library. After sequence analysis, copy number variation (CNV) has estimated using PopSV 1.0.0 R package. The RF algorithm and Gaussian Mclust model applied using gene bodies and DhMRs for cancer type prediction with different cancer stages from forty HCC, pancreatic lung cancer patients, and healthy samples. RF algorithm achieved the highest accuracy, 87.5% and 92%, for two feature sets, gene bodies, and DhMRs, while Mclust prediction accuracies are 82.5% and 90%.
Cohen et al. [
170] designed CancerSEEK method for early cancer detection using circulating protein biomarkers and mutation in cfDNA from multi-analyte blood test results consist of 1817 blood plasma samples with 1005 cancer patients with eight different type of cancer such as colorectum, liver, ovary, esophagus, pancreas, stomach, breast and lung cancer, and 812 healthy individuals. CancerSEEK method is usually applied for both binary and localize cancer detection from the mentioned blood test. For binary cancer detection, logistic regression (LR) classifier with 10-folds cross-validation involved using omega cfDNA score and eight protein biomarkers. CancerSEEK employed the random forest (RF) classifier with 10-folds cross-validation using omega cfDNA score, 39 protein biomarkers, and patient gender for cancer type localization. CancerSEEK achieved 70% average sensitivity for eight cancer types with 99% specificity, including the sensitivity levels of five cancer types from 69% to 98%.
Later, Nassri et al. [
172] applied the binomial RF classifier for gliomas cancer detection with other types of cancer using cfDNA methylation profile from plasma samples. They achieved the highest sensitivity with an AUC value of 0.990.
Penson et al. [
171] used the RF machine learning classifier for cancer type detection on tissue biopsies and then validated on two plasma ctDNA datasets. They achieved 73.8% accuracy with 5-folds cross-validation for 22 cancer types, including the highest accuracy of 95%, 87%, and 85% for uveal melanoma, glioma, and colorectal cancer, respectively. It also obtained 75% accuracy from plasma ctDNA genome analysis.
Wang et al. [
176] used the RF model for gastrointestinal cancer detection using plasma cfDNA data. The gastrointestinal cancers include the gall bladder, stomach, esophagus, colon, bile duct, pancreas, liver, and rectum cancers. This study also analyzed the cfDNA profile of hepatocellular carcinoma, colorectal cancer, pancreatic cancer patients, and healthy individuals. It obtained the AUC of 0.960, 0.890, 0.910, respectively, using the RF model with 10-folds cross-validation.
Zhang et al. [
173] employed the RF algorithm for feature selection and classification of early-stage lung cancer using circulating miRNA from the liquid biopsy with SMOTE oversampling technique. They achieved the highest 96.60% accuracy value (AUC = 0.996) with a maximum of 13 miRNA features. RF identified the top five circulating miRNA features for early lung cancer detection.
Peng et al. [
177] applied the RF prediction model for early-stage pancreatic cancer detection of diabetic patients using blood-based plasma biomarkers. The RF model has identified the best biomarkers for early-stage pancreatic cancer patients considering the AUC measure using the leave-one-out cross-validation technique and obtained AUC values of 0.850 and 0.810 with and without the CA19-9 biomarker.
Hoshino et al. [
174] employed the RF classifier to identified the biomarkers from extracellular vesicles and particle (EVP) for cancer detection. The research shows that EVP proteins are able to serve as biomarkers for early cancer detection and tumor origin detection. This study used 426 human EVP profile samples for cancer detection and achieved over 90% sensitivity and 88% specificity on both training set and test set.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}