
Evolving Mechanisms in the Pathophysiology of Pemphigus Vulgaris: A Review Emphasizing the Role of Desmoglein 3 in Regulating p53 and the Yes-Associated Protein


Abstract
1. Introduction
2. Pathogenesis of PV
2.1. Steric Hindrance of Cell Adhesion Caused by Autoantibodies
2.2. Desmoglein Compensation
2.3. Outside-In Signaling
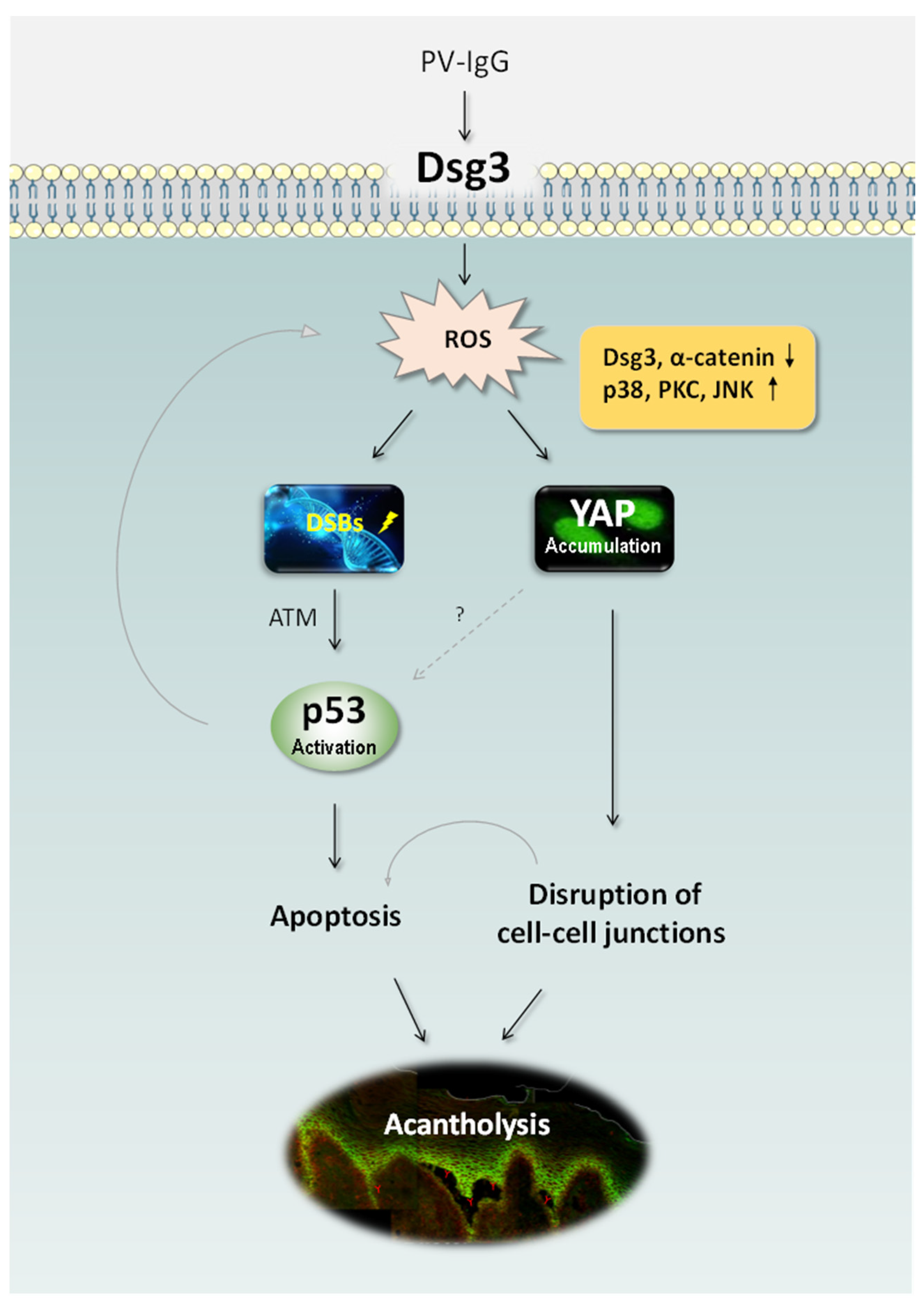
2.4. Antibody-Induced Cell Shrinkage and Apoptosis Involved in the Activation of the p53 Pathway
2.5. Oxidative Stress-Mediated YAP Dysregulation

2.6. Non-Desmoglein Antibodies
2.7. T-Cell Dysregulation
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Saito, M.; Stahley, S.N.; Caughman, C.Y.; Mao, X.; Tucker, D.K.; Payne, A.S.; Amagai, M.; Kowalczyk, A.P. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS ONE 2012, 7, e50696. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M.; Koch, P.J.; Nishikawa, T.; Stanley, J.R. Pemphigus vulgaris antigen (desmoglein 3) is localized in the lower epidermis, the site of blister formation in patients. J. Investig. Dermatol. 1996, 106, 351–355. [Google Scholar] [CrossRef]
- Amagai, M.; Tsunoda, K.; Zillikens, D.; Nagai, T.; Nishikawa, T. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J. Am. Acad. Dermatol. 1999, 40, 167–170. [Google Scholar] [CrossRef]
- Karpati, S.; Amagai, M.; Prussick, R.; Cehrs, K.; Stanley, J.R. Pemphigus vulgaris antigen, a desmoglein type of cadherin, is localized within keratinocyte desmosomes. J. Cell Biol. 1993, 122, 409–415. [Google Scholar] [CrossRef]
- Amagai, M.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991, 67, 869–877. [Google Scholar] [CrossRef]
- Anhalt, G.J.; Labib, R.S.; Voorhees, J.J.; Beals, T.F.; Diaz, L.A. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N. Engl. J. Med. 1982, 306, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Vielmuth, F.; Wanuske, M.T.; Seifert, M.; Pollmann, R.; Eming, R.; Waschke, J. Role of Dsg1- and Dsg3-Mediated Signaling in Pemphigus Autoantibody-Induced Loss of Keratinocyte Cohesion. Front. Immunol. 2019, 10, 1128. [Google Scholar] [CrossRef]
- Kitajima, Y. New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J. Med. Sci. 2013, 29, 1–13. [Google Scholar] [CrossRef]
- Sajda, T.; Sinha, A.A. Autoantibody Signaling in Pemphigus Vulgaris: Development of an Integrated Model. Front. Immunol. 2018, 9, 692. [Google Scholar] [CrossRef]
- Spindler, V.; Eming, R.; Schmidt, E.; Amagai, M.; Grando, S.; Jonkman, M.F.; Kowalczyk, A.P.; Muller, E.J.; Payne, A.S.; Pincelli, C.; et al. Mechanisms Causing Loss of Keratinocyte Cohesion in Pemphigus. J. Investig. Dermatol. 2018, 138, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.M.; Brown, L.; Lin, K.; Liu, L.; Piper, K.; O’Toole, E.A.; Grose, R.; Hart, I.R.; Garrod, D.R.; Fortune, F.; et al. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J. Pathol. 2012, 227, 81–93. [Google Scholar] [CrossRef]
- Merritt, A.J.; Berika, M.Y.; Zhai, W.; Kirk, S.E.; Ji, B.; Hardman, M.J.; Garrod, D.R. Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Mol. Cell. Biol. 2002, 22, 5846–5858. [Google Scholar] [CrossRef] [PubMed]
- Mannan, T.; Jing, S.; Foroushania, S.H.; Fortune, F.; Wan, H. RNAi-mediated inhibition of the desmosomal cadherin (desmoglein 3) impairs epithelial cell proliferation. Cell Prolif. 2011, 44, 301–310. [Google Scholar] [CrossRef]
- Chen, Y.J.; Lee, L.Y.; Chao, Y.K.; Chang, J.T.; Lu, Y.C.; Li, H.F.; Chiu, C.C.; Li, Y.C.; Li, Y.L.; Chiou, J.F.; et al. DSG3 facilitates cancer cell growth and invasion through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP signaling pathway. PLoS ONE 2013, 8, e64088. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Cai, Y.; Hünefeld, C.; Jedličková, H.; Huang, Y.; Teck Teh, M.; Sharif Ahmad, U.; Uttagomol, J.; Wang, Y.; Kang, A.; et al. The desmosomal cadherin desmoglein-3 acts as a keratinocyte anti-stress protein via suppression of p53. Cell Death Dis. 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, Y.; Kitajima, Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J. Investig. Dermatol. 1999, 112, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.M.; Brown, L.; Gadmor, H.; Gammon, L.; Fortune, F.; Wheeler, A.; Wan, H. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp. Cell Res. 2012, 318, 2269–2283. [Google Scholar] [CrossRef]
- Tsang, S.M.; Liu, L.; Teh, M.T.; Wheeler, A.; Grose, R.; Hart, I.R.; Garrod, D.R.; Fortune, F.; Wan, H. Desmoglein 3, via an interaction with E-cadherin, is associated with activation of Src. PLoS ONE 2010, 5, e14211. [Google Scholar] [CrossRef]
- Brown, L.; Wan, H. Desmoglein 3: A help or a hindrance in cancer progression? Cancers 2015, 7, 266–286. [Google Scholar] [CrossRef]
- Amagai, M.; Ahmed, A.R.; Kitajima, Y.; Bystryn, J.C.; Milner, Y.; Gniadecki, R.; Hertl, M.; Pincelli, C.; Kurzen, H.; Fridkis-Hareli, M.; et al. Are desmoglein autoantibodies essential for the immunopathogenesis of pemphigus vulgaris, or just “witnesses of disease”? Exp. Dermatol. 2006, 15, 815–831. [Google Scholar] [CrossRef]
- Grando, S.A.; Bystryn, J.C.; Chernyavsky, A.I.; Frusic-Zlotkin, M.; Gniadecki, R.; Lotti, R.; Milner, Y.; Pittelkow, M.R.; Pincelli, C. Apoptolysis: A novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 2009, 18, 764–770. [Google Scholar] [CrossRef]
- Lanza, A.; Cirillo, N.; Femiano, F.; Gombos, F. How does acantholysis occur in pemphigus vulgaris: A critical review. J. Cutan. Pathol. 2006, 33, 401–412. [Google Scholar] [CrossRef]
- Spindler, V.; Waschke, J. Pemphigus-A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front. Immunol. 2018, 9, 136. [Google Scholar] [CrossRef]
- Kitajima, Y. 150(th) anniversary series: Desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun. Adhes. 2014, 21, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, K.; Ota, T.; Aoki, M.; Yamada, T.; Nagai, T.; Nakagawa, T.; Koyasu, S.; Nishikawa, T.; Amagai, M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J. Immunol. 2003, 170, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Hammers, C.M.; Stanley, J.R. Recent Advances in Understanding Pemphigus and Bullous Pemphigoid. J Invest Dermatol. 2020, 140, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.A.; Sajda, T. The Evolving Story of Autoantibodies in Pemphigus Vulgaris: Development of the “Super Compensation Hypothesis”. Front. Med. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Sardana, K.; Garg, V.K.; Agarwal, P. Is there an emergent need to modify the desmoglein compensation theory in pemphigus on the basis of Dsg ELISA data and alternative pathogenic mechanisms? Br. J. Dermatol. 2013, 168, 669–674. [Google Scholar] [CrossRef]
- Aoyama, Y.; Owada, M.K.; Kitajima, Y. A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur. J. Immunol. 1999, 29, 2233–2240. [Google Scholar] [CrossRef]
- Osada, K.; Seishima, M.; Kitajima, Y. Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J. Investig. Dermatol. 1997, 108, 482–487. [Google Scholar] [CrossRef]
- Bektas, M.; Jolly, P.S.; Berkowitz, P.; Amagai, M.; Rubenstein, D.S. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J. Biol. Chem. 2013, 288, 9447–9456. [Google Scholar] [CrossRef]
- Thomason, H.A.; Cooper, N.H.; Ansell, D.M.; Chiu, M.; Merrit, A.J.; Hardman, M.J.; Garrod, D.R. Direct evidence that PKCα positively regulates wound re-epithelialization: Correlation with changes in desmosomal adhesiveness. J. Pathol. 2012, 227, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Wallis, S.; Lloyd, S.; Wise, I.; Ireland, G.; Fleming, T.P.; Garrod, D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol. Biol. Cell 2000, 11, 1077–1092. [Google Scholar] [CrossRef]
- Rötzer, V.; Hartlieb, E.; Vielmuth, F.; Gliem, M.; Spindler, V.; Waschke, J. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell. Mol. Life Sci. 2015, 72, 4885–4897. [Google Scholar] [CrossRef] [PubMed]
- Nekrasova, O.; Green, K.J. Desmosome assembly and dynamics. Trends Cell Biol 2013, 23, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bregegere, F.; Frusic-Zlotkin, M.; Feinmesser, M.; Michel, B.; Milner, Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis 2004, 9, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Deyhimi, P.; Tavakoli, P. Study of apoptosis in oral pemphigus vulgaris using immunohistochemical marker Bax and TUNEL technique. J. Oral Pathol. Med. 2013, 42, 409–414. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Karaouni, A.; Grando, S.A. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am. J. Pathol. 2005, 167, 1531–1544. [Google Scholar] [CrossRef]
- Pelacho, B.; Natal, C.; Espana, A.; Sanchez-Carpintero, I.; Iraburu, M.J.; Lopez-Zabalza, M.J. Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett. 2004, 566, 6–10. [Google Scholar] [CrossRef]
- Pacheco-Tovar, M.G.; Avalos-Díaz, E.; Vega-Memije, E.; Bollain-y-Goytia, J.J.; López-Robles, E.; Hojyo-Tomoka, M.T.; Domínguez-Soto, L.; Herrera-Esparza, R. The final destiny of acantholytic cells in pemphigus is Fas mediated. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 697–701. [Google Scholar] [CrossRef]
- Sanath, A.K.; Devy, A.S.; Aithal, S.; Kumar, G.S.; Prasad, B.G.; Pradeep, P.S. Caspase cascade pathways of apoptosis in oral pemphigus: An immunohistochemical study. J. Oral Maxillofac. Pathol. 2018, 22, 48–53. [Google Scholar] [CrossRef]
- Mario, P. Fas Ligand in Pemphigus Sera Induces Keratinocyte Apoptosis through the Activation of Caspase-8. J. Investig. Dermatol. 2003, 120, 164–167. [Google Scholar]
- Schmidt, E.; Waschke, J. Apoptosis in pemphigus. Autoimmun. Rev. 2009, 8, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Pemphigus autoimmunity: Hypotheses and realities. Autoimmunity 2012, 45, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.; Hunziker, T.; Suter, M.M.; Muller, E.J. Nuclear c-Myc: A molecular marker for early stage pemphigus vulgaris. J. Investig. Dermatol. 2007, 127, 1549–1555. [Google Scholar] [CrossRef]
- Williamson, L.; Raess, N.A.; Caldelari, R.; Zakher, A.; de Bruin, A.; Posthaus, H.; Bolli, R.; Hunziker, T.; Suter, M.M.; Muller, E.J. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. Embo J. 2006, 25, 3298–3309. [Google Scholar] [CrossRef]
- Chen, Y.; Miao, Z.H.; Zhao, W.M.; Ding, J. The p53 pathway is synergized by p38 MAPK signaling to mediate 11,11’-dideoxyverticillin-induced G2/M arrest. FEBS Lett. 2005, 579, 3683–3690. [Google Scholar] [CrossRef]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef]
- Lee, H.E.; Berkowitz, P.; Jolly, P.S.; Diaz, L.A.; Chua, M.P.; Rubenstein, D.S. Biphasic Activation of p38MAPK Suggests That Apoptosis Is a Downstream Event in Pemphigus Acantholysis. J. Biol. Chem. 2009, 284, 12524–12532. [Google Scholar] [CrossRef]
- Mavropoulos, A.; Orfanidou, T.; Liaskos, C.; Smyk, D.S.; Spyrou, V.; Sakkas, L.I.; Rigopoulou, E.I.; Bogdanos, D.P. p38 MAPK Signaling in Pemphigus: Implications for Skin Autoimmunity. Autoimmune Dis. 2013, 2013. [Google Scholar] [CrossRef]
- Shimizu, A.; Ishiko, A.; Ota, T.; Tsunoda, K.; Amagai, M.; Nishikawa, T. IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J. Investig. Dermatol. 2004, 122, 1145–1153. [Google Scholar] [CrossRef]
- Schulze, K.; Galichet, A.; Sayar, B.S.; Scothern, A.; Howald, D.; Zymann, H.; Siffert, M.; Zenhäusern, D.; Bolli, R.; Koch, P.J.; et al. An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J. Investig. Dermatol. 2012, 132, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Phesse, T.J.; Myant, K.B.; Cole, A.M.; Ridgway, R.A.; Pearson, H.; Muncan, V.; van den Brink, G.R.; Vousden, K.H.; Sears, R.; Vassilev, L.T.; et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014, 21, 956–966. [Google Scholar] [CrossRef]
- Pusapati, R.V.; Rounbehler, R.J.; Hong, S.; Powers, J.T.; Yan, M.; Kiguchi, K.; McArthur, M.J.; Wong, P.K.; Johnson, D.G. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Proc. Natl. Acad. Sci. USA 2006, 103, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, M.; Zillikens, D. The pathophysiology of bullous pemphigoid. Clin. Rev. Allergy Immunol. 2007, 33, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.R.; Hawley-Nelson, P.; Yuspa, S.H.; Shevach, E.M.; Katz, S.I. Characterization of bullous pemphigoid antigen: A unique basement membrane protein of stratified squamous epithelia. Cell 1981, 24, 897–903. [Google Scholar] [CrossRef]
- Hammers, C.M.; Stanley, J.R. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu. Rev. Pathol. 2016, 11, 175–197. [Google Scholar] [CrossRef]
- Tie, D.; Da, X.; Natsuga, K.; Yamada, N.; Yamamoto, O.; Morita, E. Bullous Pemphigoid IgG Induces Cell Dysfunction and Enhances the Motility of Epidermal Keratinocytes via Rac1/Proteasome Activation. Front. Immunol. 2019, 10, 200. [Google Scholar] [CrossRef]
- Wang, L.; Liu, T.; Wang, Y.; Cao, L.; Nishioka, M.; Aguirre, R.L.; Ishikawa, A.; Geng, L.; Okada, N. Altered expression of desmocollin 3, desmoglein 3, and beta-catenin in oral squamous cell carcinoma: Correlation with lymph node metastasis and cell proliferation. Virchows Arch. 2007, 451, 959–966. [Google Scholar] [CrossRef]
- Nei, H.; Saito, T.; Tobioka, H.; Itoh, E.; Mori, M.; Kudo, R. Expression of component desmosomal proteins in uterine endometrial carcinoma and their relation to cellular differentiation. Cancer 1996, 78, 461–470. [Google Scholar] [CrossRef]
- Xin, Z.; Yamaguchi, A.; Sakamoto, K. Aberrant expression and altered cellular localization of desmosomal and hemidesmosomal proteins are associated with aggressive clinicopathological features of oral squamous cell carcinoma. Virchows Arch. 2014, 465, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Pietkiewicz, P.; Gornowicz-Porowska, J.; Bowszyc-Dmochowska, M.; Jagielska, J.; Helak-Łapaj, C.; Kaczmarek, E.; Dmochowski, M. Discordant expression of desmoglein 2 and 3 at the mRNA and protein levels in nodular and superficial basal cell carcinoma revealed by immunohistochemistry and fluorescent in situ hybridization. Clin. Exp. Dermatol. 2014, 39, 628–635. [Google Scholar] [CrossRef]
- Gornowicz-Porowska, J.; Bowszyc-Dmochowska, M.; Seraszek-Jaros, A.; Kaczmarek, E.; Dmochowski, M. Loss of correlation between intensities of desmoglein 2 and desmoglein 3 expression in basal cell carcinomas. Acta Dermatovenerol. Croat. 2011, 19, 150–155. [Google Scholar]
- Brown, L.; Waseem, A.; Cruz, I.N.; Szary, J.; Gunic, E.; Mannan, T.; Unadkat, M.; Yang, M.; Valderrama, F.; O’Toole, E.A.; et al. Desmoglein 3 promotes cancer cell migration and invasion by regulating activator protein 1 and protein kinase C-dependent-Ezrin activation. Oncogene 2014, 33, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chang, J.T.; Lee, L.; Wang, H.M.; Liao, C.T.; Chiu, C.C.; Chen, P.J.; Cheng, A.J. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene 2007, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J. Role of free radicals and antioxidants in health and disease. Cell. Mol. Biol. 2007, 53, 1–2. [Google Scholar]
- Gasperlin, M.; Gosenca, M. Main approaches for delivering antioxidant vitamins through the skin to prevent skin ageing. Expert Opin. Drug Deliv. 2011, 8, 905–919. [Google Scholar] [CrossRef]
- Portugal, M.; Barak, V.; Ginsburg, I.; Kohen, R. Interplay among oxidants, antioxidants, and cytokines in skin disorders: Present status and future considerations. Biomed. Pharmacother. 2007, 61, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, E.; Liang, X.M.; Goda, M.; Otani, H.; Mune, M. The efficacy of vitamin E against oxidative damage and autoantibody production in systemic lupus erythematosus: A preliminary study. Clin. Rheumatol. 2007, 26, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Heliovaara, M.; Knekt, P.; Aho, K.; Aaran, R.K.; Alfthan, G.; Aromaa, A. Serum antioxidants and risk of rheumatoid arthritis. Ann. Rheumat. Dis. 1994, 53, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Karatas, F.; Ozates, I.; Canatan, H.; Halifeoglu, I.; Karatepe, M.; Colakt, R. Antioxidant status & lipid peroxidation in patients with rheumatoid arthritis. Indian J. Med. Res. 2003, 118, 178–181. [Google Scholar] [PubMed]
- Taysi, S.; Polat, F.; Gul, M.; Sari, R.A.; Bakan, E. Lipid peroxidation, some extracellular antioxidants, and antioxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol. Int. 2002, 21, 200–204. [Google Scholar] [CrossRef]
- Jaswal, S.; Mehta, H.C.; Sood, A.K.; Kaur, J. Antioxidant status in rheumatoid arthritis and role of antioxidant therapy. Clin. Chim. Acta Int. J. Clin. Chem. 2003, 338, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.Z.; Gheita, T.A.; Kenawy, S.A.; Fahim, A.T.; El-Sorougy, I.M.; Abdou, M.S. Oxidative stress in systemic lupus erythematosus and rheumatoid arthritis patients: Relationship to disease manifestations and activity. Int. J. Rheumat. Dis. 2011, 14, 325–331. [Google Scholar] [CrossRef]
- Naziroglu, M.; Kokcam, I.; Simsek, H.; Karakilcik, A.Z. Lipid peroxidation and antioxidants in plasma and red blood cells from patients with pemphigus vulgaris. J. Basic Clin. Physiol. Pharmacol. 2003, 14, 31–42. [Google Scholar] [CrossRef]
- Yesilova, Y.; Ucmak, D.; Selek, S.; Dertlioglu, S.B.; Sula, B.; Bozkus, F.; Turan, E. Oxidative stress index may play a key role in patients with pemphigus vulgaris. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, M.; Rahimi, H.; Barikbin, B.; Toossi, P.; Lotfi, S.; Hedayati, M.; Younespour, S. Uric Acid: A new antioxidant in patients with pemphigus vulgaris. Indian J. Dermatol. 2011, 56, 278–281. [Google Scholar] [CrossRef]
- Ahmadreza, T.; Hossein, T.M.; Kafami, K.Z.; Bita, K.; Pouran, L.; Isaac, H.S. Serum protein carbonyl and total antioxidant capacity levels. Iran. J. Dermatol. 2015, 18, 156–162. [Google Scholar]
- Shah, A.A.; Dey-Rao, R.; Seiffert-Sinha, K.; Sinha, A.A. Increased oxidative stress in pemphigus vulgaris is related to disease activity and HLA-association. Autoimmunity 2016, 49, 248–257. [Google Scholar] [CrossRef]
- Javanbakht, M.H.; Djalali, M.; Daneshpazhooh, M.; Zarei, M.; Eshraghian, M.R.; Derakhshanian, H.; Chams-Davatchi, C. Evaluation of antioxidant enzyme activity and antioxidant capacity in patients with newly diagnosed pemphigus vulgaris. Clin. Exp. Dermatol. 2015, 40, 313–317. [Google Scholar] [CrossRef]
- Huang, Y.; Jedlickova, H.; Cai, Y.; Rehman, A.; Gammon, L.; Ahmad, U.S.; Uttagomol, J.; Parkinson, E.K.; Fortune, F.; Wan, H. Oxidative Stress-Mediated YAP Dysregulation Contributes to the Pathogenesis of Pemphigus Vulgaris. Front. Immunol. 2021, 12, 649502. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.C.; Moroishi, T.; Meng, Z.; Jeong, H.S.; Plouffe, S.W.; Sekido, Y.; Han, J.; Park, H.W.; Guan, K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 2017, 19, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Meng, Z.; Moroishi, T.; Lin, K.C.; Shen, G.; Mo, F.; Shao, B.; Wei, X.; Zhang, P.; Wei, Y.; et al. Publisher Correction: Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat. Cell Biol. 2021, 23, 209. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef]
- Uttagomol, J.; Ahmad, U.S.; Rehman, A.; Huang, Y.; Laly, A.C.; Kang, A.; Soetaert, J.; Chance, R.; Teh, M.T.; Connelly, J.T.; et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces. Int. J. Mol. Sci. 2019, 20, 6221. [Google Scholar] [CrossRef]
- Rehman, A.; Wan, H. A Novel Regulatory Pathway of Desmoglein-3 in Keratinocyte Stress Response. J. Cell. Signal. 2020, 1, 10. [Google Scholar]
- Vu, T.N.; Lee, T.X.; Ndoye, A.; Shultz, L.D.; Pittelkow, M.R.; Dahl, M.V.; Lynch, P.J.; Grando, S.A. The Pathophysiological Significance of Nondesmoglein Targets of Pemphigus Autoimmunity. Development of Antibodies Against Keratinocyte Cholinergic Receptors in Patients with Pemphigus Vulgaris and Pemphigus Foliaceus. Arch. Dermatol. 1998, 134. [Google Scholar] [CrossRef]
- Marchenko, S.; Chernyavsky, A.I.; Arredondo, J.; Gindi, V.; Grando, S.A. Antimitochondrial autoantibodies in pemphigus vulgaris: A missing link in disease pathophysiology. J. Biol. Chem. 2010, 285, 3695–3704. [Google Scholar] [CrossRef]
- Yin, T.; Getsios, S.; Caldelari, R.; Godsel, L.M.; Kowalczyk, A.P.; Müller, E.J.; Green, K.J. Mechanisms of plakoglobin-dependent adhesion: Desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 40355–40363. [Google Scholar] [CrossRef]
- Kalantari-Dehaghi, M.; Anhalt, G.J.; Camilleri, M.J.; Chernyavsky, A.I.; Chun, S.; Felgner, P.L.; Jasinskas, A.; Leiferman, K.M.; Liang, L.; Marchenko, S.; et al. Pemphigus Vulgaris Autoantibody Profiling by Proteomic Technique. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Sklyarova, T.; Bonné, S.; D’Hooge, P.; Denecker, G.; Goossens, S.; De Rycke, R.; Borgonie, G.; Bösl, M.; van Roy, F.; van Hengel, J. Plakophilin-3-deficient mice develop hair coat abnormalities and are prone to cutaneous inflammation. J. Investig. Dermatol. 2008, 128, 1375–1385. [Google Scholar] [CrossRef]
- Spindler, V.; Dehner, C.; Hübner, S.; Waschke, J. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J. Investig. Dermatol. 2014, 134, 1655–1664. [Google Scholar] [CrossRef]
- Korman, N.J.; Eyre, R.W.; Klaus-Kovtun, V.; Stanley, J.R. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N. Engl. J. Med. 1989, 321, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, F.; Dasher, D.A.; Diaz, L.A.; Prisayanh, P.S.; Li, N. E-cadherin is an additional immunological target for pemphigus autoantibodies. J. Investig. Dermatol. 2008, 128, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Kljuic, A.; Bazzi, H.; Sundberg, J.P.; Martinez-Mir, A.; O’Shaughnessy, R.; Mahoney, M.G.; Levy, M.; Montagutelli, X.; Ahmad, W.; Aita, V.M.; et al. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: Evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell 2003, 113, 249–260. [Google Scholar] [CrossRef]
- Nagasaka, T.; Nishifuji, K.; Ota, T.; Whittock, N.V.; Amagai, M. Defining the pathogenic involvement of desmoglein 4 in pemphigus and staphylococcal scalded skin syndrome. J. Clin. Investig. 2004, 114, 1484–1492. [Google Scholar] [CrossRef]
- Kim, S.C.; Chung, Y.L.; Kim, J.; Cho, N.J.; Amagai, M. Pemphigus vulgaris with autoantibodies to desmoplakin. Br. J. Dermatol. 2001, 145, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Ndoye, A.; Grando, S.A. Pemphigus vulgaris antibody identifies pemphaxin. A novel keratinocyte annexin-like molecule binding acetylcholine. J. Biol. Chem. 2000, 275, 29466–29476. [Google Scholar] [CrossRef]
- Gualtieri, B.; Marzano, V.; Grando, S.A. Atypical pemphigus: Autoimmunity against desmocollins and other non-desmoglein autoantigens. Ital. J. Dermatol. Venerol. 2021, 156, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.; Amber, K.T.; Agnoletti, A.F.; Wang, C.; Grando, S.A. Synergy among non-desmoglein antibodies contributes to the immunopathology of desmoglein antibody-negative pemphigus vulgaris. J. Biol. Chem. 2019, 294, 4520–4528. [Google Scholar] [CrossRef]
- Oliveira, M.E.; Culton, D.A.; Prisayanh, P.; Qaqish, B.F.; Diaz, L.A. E-cadherin autoantibody profile in patients with pemphigus vulgaris. Br. J. Dermatol. 2013, 169, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, J.M.; Zeng, K. Association between HLA-DRB1 polymorphisms and pemphigus vulgaris: A meta-analysis. Br. J. Dermatol. 2012, 167, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Vodo, D.; Sarig, O.; Sprecher, E. The Genetics of Pemphigus Vulgaris. Front. Med. 2018, 5, 226. [Google Scholar] [CrossRef]
- Loiseau, P.; Lecleach, L.; Prost, C.; Lepage, V.; Busson, M.; Bastuji-Garin, S.; Roujeau, J.C.; Charron, D. HLA class II polymorphism contributes to specify desmoglein derived peptides in pemphigus vulgaris and pemphigus foliaceus. J. Autoimmun. 2000, 15, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Shams, S.; Amirzargar, A.A.; Yousefi, M.; Rezaei, N.; Solgi, G.; Khosravi, F.; Ansaripour, B.; Moradi, B.; Nikbin, B. HLA class II (DRB, DQA1 and DQB1) allele and haplotype frequencies in the patients with pemphigus vulgaris. J. Clin. Immunol. 2009, 29, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Satyam, A.; Khandpur, S.; Sharma, V.K.; Sharma, A. Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-Pemphigus vulgaris. Immunol. Investig. 2009, 38, 498–509. [Google Scholar] [CrossRef]
- Arakawa, M.; Dainichi, T.; Yasumoto, S.; Hashimoto, T. Lesional Th17 cells in pemphigus vulgaris and pemphigus foliaceus. J. Dermatol. Sci. 2009, 53, 228–231. [Google Scholar] [CrossRef]
- Xu, R.C.; Zhu, H.Q.; Li, W.P.; Zhao, X.Q.; Yuan, H.J.; Zheng, J.; Pan, M. The imbalance of Th17 and regulatory T cells in pemphigus patients. Eur. J. Dermatol. 2013, 23, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Asothai, R.; Anand, V.; Das, D.; Antil, P.S.; Khandpur, S.; Sharma, V.K.; Sharma, A. Distinctive Treg associated CCR4-CCL22 expression profile with altered frequency of Th17/Treg cell in the immunopathogenesis of Pemphigus Vulgaris. Immunobiology 2015, 220, 1129–1135. [Google Scholar] [CrossRef]
- Yokoyama, T.; Matsuda, S.; Takae, Y.; Wada, N.; Nishikawa, T.; Amagai, M.; Koyasu, S. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int. Immunol. 2011, 23, 365–373. [Google Scholar] [CrossRef]
- Schmidt, T.; Willenborg, S.; Hünig, T.; Deeg, C.A.; Sonderstrup, G.; Hertl, M.; Eming, R. Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp. Dermatol. 2016, 25, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ahmad, U.S.; Huang, Y.; Uttagomol, J.; Rehman, A.; Zhou, K.; Warnes, G.; McArthur, S.; Parkinson, E.K.; Wan, H. Desmoglein-3 acts as a pro-survival protein by suppressing reactive oxygen species and doming whilst augmenting the tight junctions in MDCK cells. Mech. Ageing Dev. 2019, 184, 111174. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Jaiganesh, A.; Broussard, J.A. Desmosomes: Essential contributors to an integrated intercellular junction network. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Origins | Sample Size | Components Detection | Oxidant/Antioxidant Imbalance | Conclusions | References |
|---|---|---|---|---|---|
| Plasma & RBC | 18 | Levels of lipid peroxidation and antioxidants | ↑ Oxidant/↓ Antioxidant (Vitamin A & E, β-carotene, CAT, GSH-Px, GSH) | Oxidant/Antioxidant imbalance predominantly in favor of pro-oxidant side, and/or diminished TAC or levels of antioxidants | [75] |
| Plasma | 30 | Levels of antioxidants | Oxidant/↓ Antioxidant (Uric acid) | [77] | |
| Serum | 27 | Levels of TOC, LOOH, TAC OSI = TOC/TAC | ↑ Oxidant (TOC, LOOH, OSI)/Antioxidant | [76] | |
| Serum | 47 | Levels of TAC | Oxidant/↓ Antioxidant (TAC) | [79] | |
| Plasma & Serum | 43 | Levels of CAT, GSH-Px, TAC | ↑ Oxidant (CAT, GSH-Px)/↓ Antioxidant (TAC) | [80] | |
| Serum | 9 | Levels of PC and TAC | ↑ Oxidant (PC)/↑ Antioxidant (TAC) | Only study shown oxidant/antioxidant imbalance with enhanced TAC | [78] |
| Keratinocytes | 8 | ROS levels in cells treated by PV sera or AK23 | ↑ Oxidant (ROS) | Oxidant/Antioxidant imbalance with ROS overproduction evoked by PV sera or AK23 | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, A.; Huang, Y.; Wan, H. Evolving Mechanisms in the Pathophysiology of Pemphigus Vulgaris: A Review Emphasizing the Role of Desmoglein 3 in Regulating p53 and the Yes-Associated Protein. Life 2021, 11, 621. https://doi.org/10.3390/life11070621
Rehman A, Huang Y, Wan H. Evolving Mechanisms in the Pathophysiology of Pemphigus Vulgaris: A Review Emphasizing the Role of Desmoglein 3 in Regulating p53 and the Yes-Associated Protein. Life. 2021; 11(7):621. https://doi.org/10.3390/life11070621
Chicago/Turabian StyleRehman, Ambreen, Yunying Huang, and Hong Wan. 2021. "Evolving Mechanisms in the Pathophysiology of Pemphigus Vulgaris: A Review Emphasizing the Role of Desmoglein 3 in Regulating p53 and the Yes-Associated Protein" Life 11, no. 7: 621. https://doi.org/10.3390/life11070621
APA StyleRehman, A., Huang, Y., & Wan, H. (2021). Evolving Mechanisms in the Pathophysiology of Pemphigus Vulgaris: A Review Emphasizing the Role of Desmoglein 3 in Regulating p53 and the Yes-Associated Protein. Life, 11(7), 621. https://doi.org/10.3390/life11070621