
New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Normal Vascular Wall
3. Vascular Wall Alterations in Systemic Sclerosis and Related Clinical Manifestations
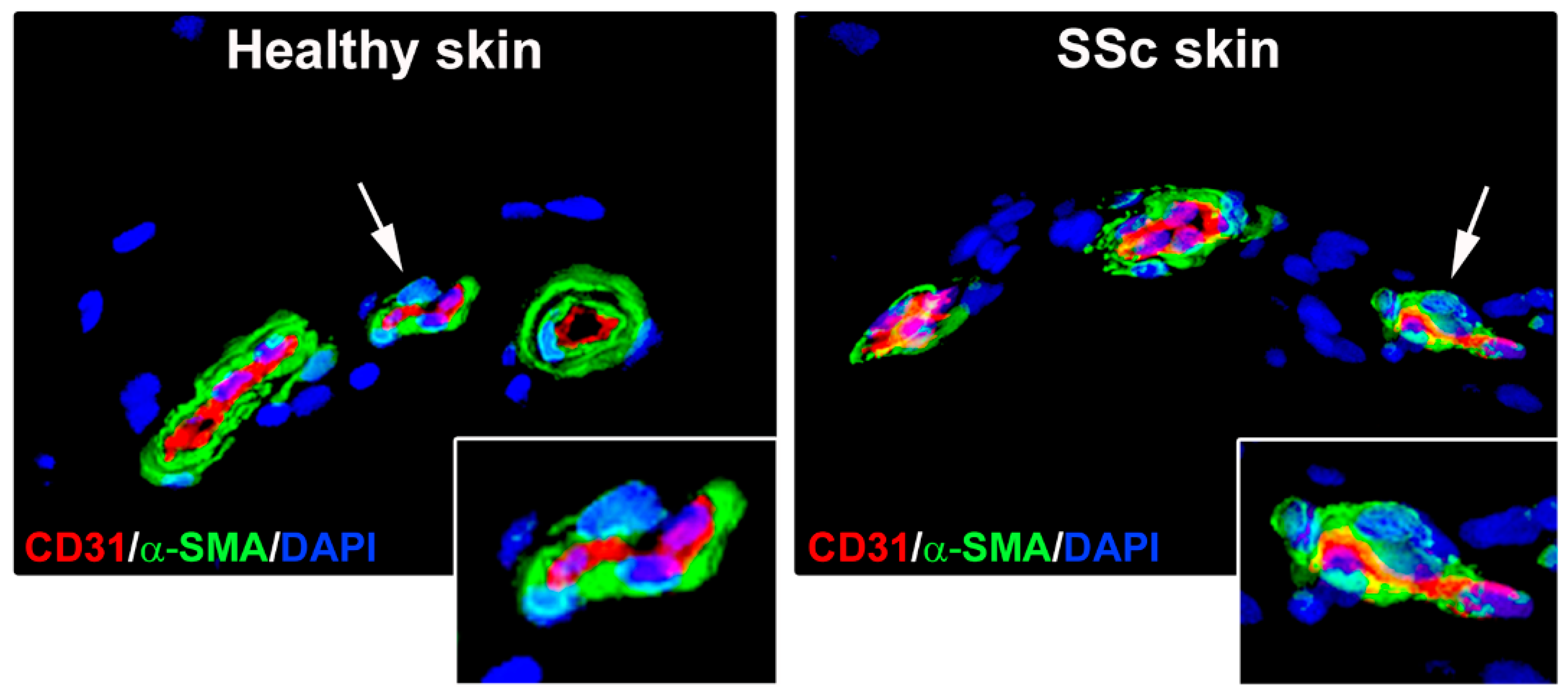
3.1. Skin
3.2. Lung
3.3. Kidney
3.4. Heart
3.5. Gastrointestinal Tract
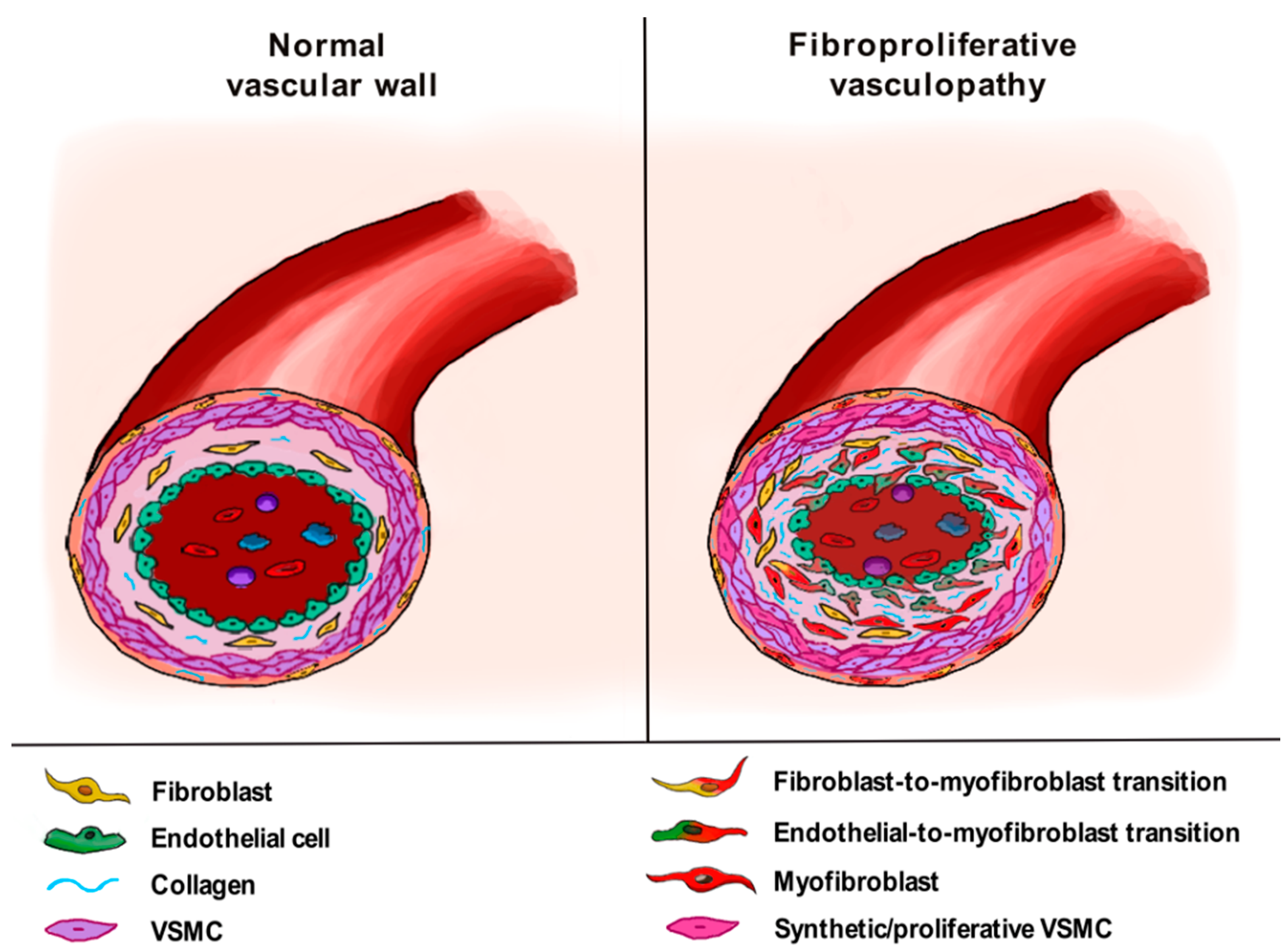
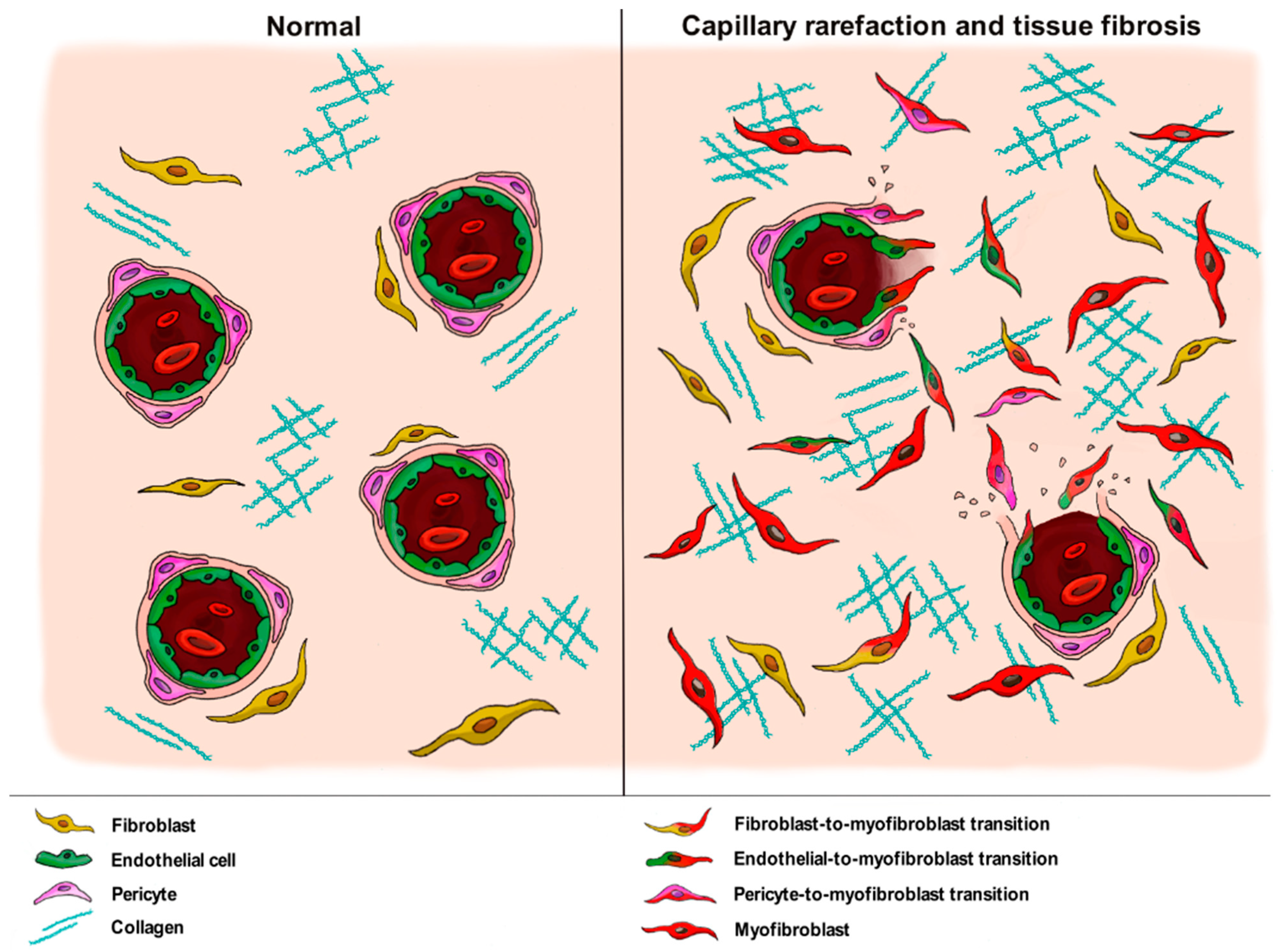
4. The Pathogenic Role of Vascular Wall-Resident Cells in Systemic Sclerosis: Linking Vasculopathy to Fibrosis
4.1. Endothelial Cells
4.2. Pericytes
4.3. Vascular Smooth Muscle Cells
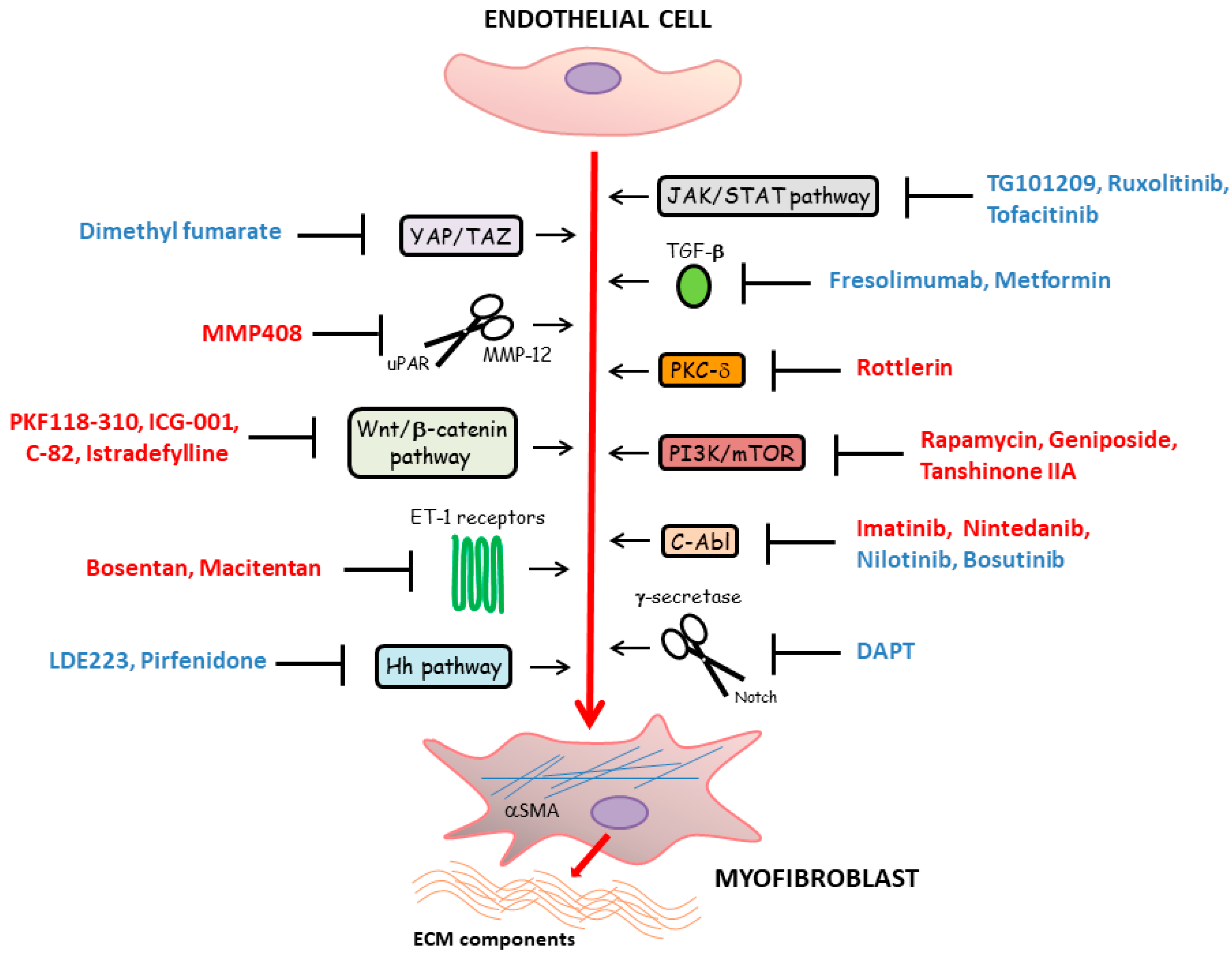
5. Main Molecular Pathways Driving Myofibroblast Differentiation from Vascular Wall Residing Cells in Systemic Sclerosis and Related Therapeutic Strategies
5.1. TGF-β Signaling
5.2. Developmental Pathways
5.3. Janus Kinase/Signal Transducers and Activators of Transcription (JAK/STAT) Signaling Pathway
5.4. Additional Therapeutic Targets
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Varga, J.; Trojanowska, M.; Kuwana, M. Pathogenesis of systemic sclerosis: Recent insights of molecular and cellular mechanisms and therapeutic opportunities. J. Scleroderma Relat. Disord. 2017, 2, 137–152. [Google Scholar] [CrossRef]
- Sierra-Sepúlveda, A.; Esquinca-González, A.; Benavides-Suárez, S.A.; Sordo-Lima, D.E.; Caballero-Islas, A.E.; Cabral-Castañeda, A.R.; Rodríguez-Reyna, T.S. Systemic sclerosis pathogenesis and emerging therapies, beyond the fibroblast. Biomed. Res. Int. 2019, 2019, 4569826. [Google Scholar] [CrossRef]
- Korman, B. Evolving insights into the cellular and molecular pathogenesis of fibrosis in systemic sclerosis. Transl. Res. 2019, 209, 77–89. [Google Scholar] [CrossRef]
- Asano, Y. The pathogenesis of systemic sclerosis: An understanding based on a common pathologic cascade across multiple organs and additional organ-specific pathologies. J. Clin. Med. 2020, 9, 2687. [Google Scholar] [CrossRef] [PubMed]
- Trojanowska, M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat. Rev. Rheumatol. 2010, 6, 453–460. [Google Scholar] [CrossRef]
- Colaci, M.; Dal Bosco, Y.; Schinocca, C.; Ronsivalle, G.; Guggino, G.; De Andres, I.; Russo, A.A.; Sambataro, D.; Sambataro, G.; Malatino, L. Aortic root dilation in associated with the reduction in capillary density observed at nailfold capillaroscopy in SSc patients. Clin. Rheumatol. 2021, 40, 1185–1189. [Google Scholar] [CrossRef]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Rosa, I.; Romano, E.; Fioretto, B.S.; Manetti, M. The contribution of mesenchymal transitions to the pathogenesis of systemic sclerosis. Eur. J. Rheumatol. 2020, 7, S157–S164. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Vomero, M.; Navarini, L.; Dolo, V.; Cipriani, P.; Giacomelli, R. Endothelial-to-mesenchymal transition in systemic sclerosis. Clin. Exp. Immunol. 2021, 205. [Google Scholar] [CrossRef] [PubMed]
- Marziano, C.; Genet, G.; Hirschi, K.K. Vascular endothelial cell specification in health and disease. Angiogenesis 2021, 24, 213. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Rigor, R.R. Structure and function of exchange microvessels. In Regulation of Endothelial Barrier Function; Granger, D.N., Granger, J., Eds.; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Milutinović, A.; Šuput, D.; Zorc-Pleskovič, R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: An updated review. Bosn. J. Basic Med. Sci. 2020, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Stratman, A.N.; Malotte, K.M.; Mahan, R.D.; Davis, M.J.; Davis, G.E. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 2009, 114, 5091–5101. [Google Scholar] [CrossRef]
- Van Dijk, C.G.; Nieuweboer, F.E.; Pei, J.Y.; Xu, Y.J.; Burgisser, P.; van Mulligen, E.; el Azzouzi, H.; Duncker, D.J.; Verhaar, M.C.; Cheng, C. The complex mural cell: Pericyte function in health and disease. Int. J. Cardiol. 2015, 190, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Mäkinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Feghali-Bostwick, C.; Varga, J. Introduction: The etiopathogenesis of systemic sclerosis—An integrated overview. In Scleroderma, 2nd ed.; Part III The Biological Basis of Systemic Sclerosis; Varga, J., Denton, C., Wigley, F.M., Allanore, Y., Kuwana, M., Eds.; Springer: New York, NY, USA, 2017; pp. 133–140. [Google Scholar] [CrossRef]
- Kavian, N.; Batteux, F. Macro- and microvascular disease in systemic sclerosis. Vascul. Pharmacol. 2015, 71, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Kahaleh, B. Mechanisms of vascular disease. In Scleroderma, 2nd ed.; Part III The Biological Basis of Systemic Sclerosis; Varga, J., Denton, C., Wigley, F.M., Allanore, Y., Kuwana, M., Eds.; Springer: New York, NY, USA, 2017; pp. 221–244. [Google Scholar] [CrossRef]
- Saygin, D.; Highland, K.B.; Tonelli, A.R. Microvascular involvement in systemic sclerosis and systemic lupus erythematosus. Microcirculation 2019, 26, e12440. [Google Scholar] [CrossRef]
- Bruni, C.; Frech, T.; Manetti, M.; Rossi, F.W.; Furst, D.E.; De Paulis, A.; Rivellese, F.; Guiducci, S.; Matucci-Cerinic, M.; Bellando-Randone, S. Vascular leaking, a pivotal and early pathogenetic event in systemic sclerosis: Should the door be closed? Front. Immunol. 2018, 9, 2045. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Vonk, M.C.; Boonstra, A.; Voskuyl, A.E.; Vonk-Noordegraaf, A.; Smit, E.F.; Dijkmans, B.A.; Postmus, P.E.; Mooi, W.J.; Heijdra, Y.; et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: A distinctive vasculopathy. Eur. Respir. J. 2009, 34, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Confalonieri, M.; Salton, F.; Wade, B.; Baratella, E.; Geri, P.; Confalonieri, P.; Kodric, M.; Biolo, M.; Bruni, C. The relationship between pulmonary damage and peripheral vascular manifestations in systemic sclerosis patients. Pharmaceuticals 2021, 14, 403. [Google Scholar] [CrossRef]
- Bruni, C.; Guignabert, C.; Manetti, M.; Matucci Cerinic, M.; Humbert, M. The multifaceted problem of pulmonary arterial hypertension in systemic sclerosis. Lancet Rheumatol. 2020, 3, e149–e159. [Google Scholar] [CrossRef]
- Rooper, L.M.; Askin, F.B. Pathology of systemic sclerosis. In Scleroderma, 2nd ed.; Part III The Biological Basis of Systemic Sclerosis; Varga, J., Denton, C., Wigley, F.M., Allanore, Y., Kuwana, M., Eds.; Springer: New York, NY, USA, 2017; pp. 141–160. [Google Scholar] [CrossRef]
- Hassoun, P.M. Lung involvement in systemic sclerosis. Presse Med. 2011, 40, e3–e17. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Humbert, M.; Perros, F.; Sanchez, O.; Simonneau, G.; Müller, K.M.; Capron, F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum. Pathol. 2007, 38, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, G.; Daoussis, D. Renal dysfunction in systemic sclerosis beyond scleroderma renal crisis. Rheumatol. Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, E.; Codullo, V.; Allanore, Y. Scleroderma renal crisis: Case reports and update on critical issues. Eur. J. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bose, N.; Chiesa-Vottero, A.; Chatterjee, S. Scleroderma renal crisis. Semin. Arthritis Rheum. 2005, 44, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Batal, I.; Domsic, R.T.; Medsger, T.A.; Bastacky, S. Scleroderma renal crisis: A pathology perspective. Int. J. Rheumatol. 2010, 2010, 543704. [Google Scholar] [CrossRef]
- Nikpour, M.; Baron, M. Mortality in systemic sclerosis: Lessons learned from population-based and observational cohort studies. Curr. Opin. Rheumatol. 2014, 26, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.; Sebastiani, M.; Lo Monaco, A.; Iudici, M.; Giuggioli, D.; Furini, F.; Manfredi, A.; Cuomo, G.; Spinella, A.; Colaci, M.; et al. Systemic sclerosis evolution of disease pathomorphosis and survival. Our experience on Italian patients’ population and review of the literature. Autoimmun. Rev. 2014, 13, 1026–1034. [Google Scholar] [CrossRef]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Bruni, C.; Tomelleri, A.; Dagna, L.; Matucci-Cerinic, M. Interleukin-1 and systemic sclerosis: Getting to the heart of cardiac involvement. Front. Immunol. 2021, 12, 653950. [Google Scholar] [CrossRef] [PubMed]
- Bruni, C.; Ross, L. Cardiac involvement in systemic sclerosis: Getting to the heart of the matter. Best Pract. Res. Clin. Rheumatol. 2021. [Google Scholar] [CrossRef]
- Allanore, Y.; Meune, C. Primary myocardial involvement in systemic sclerosis: Evidence for a microvascular origin. Clin. Exp. Rheumatol. 2010, 28, S48–S53. [Google Scholar]
- Lambova, S. Cardiac manifestations in systemic sclerosis. World J. Cardiol. 2014, 6, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Shoenfeld, Y. Atherosclerosis and macrovascular involvement in systemic sclerosis: Myth or reality. Autoimmun. Rev. 2011, 10, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Klein-Weigel, P.; Opitz, C.; Riemekasten, G. Systemic sclerosis—A systematic overview: Part 1—Disease characteristics and classification, pathophysiologic concepts, and recommendations for diagnosis and surveillance. VASA 2011, 40, 6–19. [Google Scholar] [CrossRef]
- Johnson, S.R.; Glaman, D.D.; Schentag, C.T.; Lee, P. Quality of life and functional status in systemic sclerosis compared to other rheumatic diseases. J. Rheumatol. 2006, 33, 1117–1122. [Google Scholar] [PubMed]
- McFarlane, I.M.; Bhamra, M.S.; Kreps, A.; Iqbal, S.; Al-Ani, F.; Saladini-Aponte, C.; Grant, C.; Singh, S.; Awwal, K.; Koci, K.; et al. Gastrointestinal manifestations of systemic sclerosis. Rheumatology (Sunnyvale) 2018, 8, 235. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.; Noviani, M.; Leung, Y.Y.; Low, A.H.L. The microbiome and systemic sclerosis: A review of current evidence. Best Pract. Res. Clin. Rheumatol. 2021. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Covelli, M.; Berardino, M.; Wang, D.Q.; Lapadula, G.; Palasciano, G.; Portincasa, P. Gastrointestinal symptoms and motility disorders in patients with systemic scleroderma. BMC Gastroenterol. 2008, 8, 7. [Google Scholar] [CrossRef]
- Romano, E.; Rosa, I.; Fioretto, B.S.; Cerinic, M.M.; Manetti, M. The role of pro-fibrotic myofibroblasts in systemic sclerosis: From origin to therapeutic targeting. Curr. Mol. Med. 2021. [Google Scholar] [CrossRef]
- Kanno, Y. The role of fibrinolytic regulators in vascular dysfunction of systemic sclerosis. Int. J. Mol. Sci. 2019, 20, 619. [Google Scholar] [CrossRef]
- Thuan, D.T.B.; Zayed, H.; Eid, A.H.; Abou-Saleh, H.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front. Immunol. 2018, 9, 1985. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Matucci-Cerinic, M. The origin of the myofibroblast in fibroproliferative vasculopathy: Does the endothelial cell steer the pathophysiology of systemic sclerosis? Arthritis Rheum. 2011, 63, 2164–2167. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef]
- Manetti, M.; Rosa, I.; Milia, A.F.; Guiducci, S.; Carmeliet, P.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: A new model of experimental scleroderma? Ann. Rheum. Dis. 2014, 73, 1700–1709. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Romano, E.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Increased serum levels and tissue expression of matrix metalloproteinase-12 in patients with systemic sclerosis: Correlation with severity of skin and pulmonary fibrosis and vascular damage. Ann. Rheum. Dis. 2012, 71, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Margheri, F.; Peppicelli, S.; Bianchini, F.; Ruzzolini, J.; Laurenzana, A.; Fibbi, G.; Bruni, C.; Bellando-Randone, S.; Guiducci, S.; et al. Glycolysis-derived acidic microenvironment as a driver of endothelial dysfunction in systemic sclerosis. Rheumatology 2021, keab022. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.A.; Tombetti, E.; Giovenzana, A.; Donè, E.; Pulcinelli, E.; Meneveri, R.; Tirone, M.; Maugeri, N.; Rovere-Querini, P.; Manfredi, A.A.; et al. Macrophages guard endothelial lineage by hindering endothelial-to-mesenchymal transition: Implications for the pathogenesis of systemic sclerosis. J. Immunol. 2019, 203, 247–258. [Google Scholar] [CrossRef]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Capece, D.; Zazzeroni, F.; Liakouli, V.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Pecetti, G.; et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J. Rheumatol. 2015, 42, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Corallo, C.; Cutolo, M.; Kahaleh, B.; Pecetti, G.; Montella, A.; Chirico, C.; Soldano, S.; Nuti, R.; Giordano, N. Bosentan and macitentan prevent the endothelial-to-mesenchymal transition (EndoMT) in systemic sclerosis: In vitro study. Arthritis Res. Ther. 2016, 18, 228. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, P.J.; Carney, K.R.; Mendoza, F.A.; Piera-Velazquez, S.; Jimenez, S.A. Endothelial cell-specific activation of transforming growth factor-β signaling in mice induces cutaneous, visceral, and microvascular fibrosis. Lab. Investig. 2017, 97, 806–818. [Google Scholar] [CrossRef]
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 deficiency impairs vascular homeostasis: A role in scleroderma vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998. [Google Scholar] [CrossRef]
- Taniguchi, T.; Asano, Y.; Akamata, K.; Noda, S.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Trojanowska, M.; Sato, S. Fibrosis, vascular activation, and immune abnormalities resembling systemic sclerosis in bleomycin-treated Fli-1-haploinsufficient mice. Arthritis Rheumatol. 2015, 67, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M. Fli1 deficiency and beyond: A unique pathway linking peripheral vasculopathy and dermal fibrosis in systemic sclerosis. Exp. Dermatol. 2015, 24, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Taniguchi, T.; Hirabayashi, M.; Yamashita, T.; Saigusa, R.; Miura, S.; Takahashi, T.; Toyama, T.; Ichimura, Y.; Yoshizaki, A.; et al. Altered properties of endothelial cells and mesenchymal stem cells underlying the development of scleroderma-like vasculopathy in KLF5+/−; Fli-1+/−mice. Arthritis Rheumatol. 2020, 72, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Marden, G.; Wan, Q.; Wilks, J.; Nevin, K.; Feeney, M.; Wisniacki, N.; Trojanowski, M.; Bujor, A.; Stawski, L.; Trojanowska, M. The role of the oncostatin M/OSM receptor β axis in activating dermal microvascular endothelial cells in systemic sclerosis. Arthritis Res. Ther. 2020, 22, 179. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Lv, J.; Jiang, Z.; He, H.; Chen, C.; Xiong, Y.; Zhu, X.; Xue, Y.; Yu, Y.; Yang, S.; et al. Promotion of myofibroblast differentiation and tissue fibrosis by the leukotriene B4-Leukotriene B4 receptor axis in systemic sclerosis. Arthritis Rheumatol. 2020, 72, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zaidi, M.; Struve, J.; Jones, D.W.; Krolikowski, J.G.; Nandedkar, S.; Lohr, N.L.; Gadicherla, A.; Pagel, P.S.; Csuka, M.E.; et al. Abnormal fibrillin-1 expression and chronic oxidative stress mediate endothelial mesenchymal transition in a murine model of systemic sclerosis. Am. J. Physiol. Cell. Physiol. 2011, 300, C550–C556. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Asano, Y.; Taniguchi, T.; Yamashita, T.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; Sugawara, K.; Tsuruta, D.; et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15136–15141. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Romano, E.; Ceccarelli, C.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ. Res. 2011, 109, e14–e26. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, C.; Manetti, M.; Rosa, I.; Romano, E.; Blagojevic, J.; Bellando-Randone, S.; Bruni, C.; Lepri, G.; Guiducci, S.; Ibba-Manneschi, L.; et al. Proangiogenic effects of soluble α-Klotho on systemic sclerosis dermal microvascular endothelial cells. Arthritis Res. Ther. 2017, 19, 27. [Google Scholar] [CrossRef]
- Del Galdo, F.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 2008, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, R.; Singh, H.; Pandey, A.; Mehta, A.; Bhardwaj, S.; Jaggi, A.S. Caveolin-1 as a critical component in the pathogenesis of lung fibrosis of different etiology: Evidences and mechanisms. Exp. Mol. Pathol. 2019, 111, 104315. [Google Scholar] [CrossRef]
- Li, S.; Yu, L.; He, A.; Liu, Q. Klotho inhibits unilateral ureteral obstruction-induced endothelial-to-mesenchymal transition via TGF-β1/Smad2/Snail1 signaling in mice. Front. Pharmacol. 2019, 10, 348. [Google Scholar] [CrossRef]
- Huang, X.; Pan, L.; Pu, H.; Wang, Y.; Zhang, X.; Li, C.; Yang, Z. Loss of caveolin-1 promotes endothelial-mesenchymal transition during sepsis: A membrane proteomic study. Int. J. Mol. Med. 2013, 32, 585–592. [Google Scholar] [CrossRef]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am. J. Pathol. 2013, 182, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Allanore, Y.; Saad, M.; Fatini, C.; Cohignac, V.; Guiducci, S.; Romano, E.; Airó, P.; Caramaschi, P.; Tinazzi, I. Evidence for caveolin-1 as a new susceptibility gene regulating tissue fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Murray, K.D.; Jou, M.F.; Hsu, S.M.; Cheng, H.J.; Huang, P.H. Sema3E/plexin-D1 mediated epithelial-to-mesenchymal transition in ovarian endometrioid cancer. PLoS ONE 2011, 6, e19396. [Google Scholar] [CrossRef]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of systemic sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016, 51, 26–36. [Google Scholar] [CrossRef]
- Mendoza, F.A.; Piera-Velazquez, S.; Farber, J.L.; Feghali-Bostwick, C.; Jiménez, S.A. Endothelial cells expressing endothelial and mesenchymal cell gene products in lung tissue from patients with systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. 2016, 68, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Addya, S.; Pomante, D.; Jimenez, S.A. Increased expression of interferon regulated and antiviral response genes in CD31+/CD102+ lung microvascular endothelial cells from systemic sclerosis patients with end-stage interstitial lung disease. Clin. Exp. Rheumatol. 2020. online ahead of print. [Google Scholar]
- Di Benedetto, P.; Ruscitti, P.; Liakouli, V.; Cipriani, P.; Giacomelli, R. The vessels contribute to fibrosis in systemic sclerosis. Isr. Med. Assoc. J. 2019, 21, 471–474. [Google Scholar]
- Hung, C.F.; Wilson, C.L.; Schnapp, L.M. Pericytes in the lung. Adv. Exp. Med. Biol. 2019, 1122, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Takakura, N. Role of intimate interactions between endothelial cells and the surrounding accessory cells in the maturation of blood vessels. J. Thromb. Haemost. 2011, 1, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.S.; Zhou, H.N.; He, S.S.; Xue, M.Y.; Li, T.; Liu, L.M. Research advances in pericyte function and their roles in diseases. Chin. J. Traumatol. 2020, 23, 89–95. [Google Scholar] [CrossRef]
- Greenhalgh, S.N.; Iredale, J.P.; Henderson, N.C. Origins of fibrosis: Pericytes take centre stage. F1000Prime Rep. 2013, 5, 37. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, F.; Tang, Y.; Xiao, J.; Li, C.; Ouyang, Y.; Hou, Y. Valproic acid regulates Ang II-induced pericyte-myofibroblast transdifferentiation via MAPK/ERK pathway. Am. J. Transl. Res. 2018, 10, 1976–1989. [Google Scholar]
- Chen, J.; Sivan, U.; Tan, S.L.; Lippo, L.; De Angelis, J.; Labella, R.; Singh, A.; Chatzis, A.; Cheuk, S.; Medhghalchi, M.; et al. High-resolution 3D imaging uncovers organ-specific vascular control of tissue aging. Sci. Adv. 2021, 7, eabd7819. [Google Scholar] [CrossRef] [PubMed]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef]
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A.; Zhang, T.; Files, D.C.; Mannava, S.; Smith, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 2014, 5, 122. [Google Scholar] [CrossRef]
- Wilson, C.L.; Stephenson, S.E.; Higuero, J.P.; Feghali-Bostwick, C.; Hung, C.F.; Schnapp, L.M. Characterization of human PDGFR-β-positive pericytes from IPF and non-IPF lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L991–L1002. [Google Scholar] [CrossRef]
- Rajkumar, V.S.; Howell, K.; Csiszar, K.; Denton, C.P.; Black, C.M.; Abraham, D.J. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res. Ther. 2005, 7, R1113–R1123. [Google Scholar] [CrossRef]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Taghavi, R.; Leask, A. Connective tissue growth factor is induced in bleomycin-induced skin scleroderma. J. Cell. Commun. Signal. 2010, 4, 25–30. [Google Scholar] [CrossRef]
- Hung, C.F.; Wilson, C.L.; Chow, Y.H.; Schnapp, L.M. Role of integrin alpha8 in murine model of lung fibrosis. PLoS ONE 2018, 13, e0197937. [Google Scholar] [CrossRef] [PubMed]
- Juniantito, V.; Izawa, T.; Yuasa, T.; Ichikawa, C.; Tanaka, M.; Kuwamura, M.; Yamate, J. Immunophenotypical analysis of myofibroblasts and mesenchymal cells in the bleomycin-induced rat scleroderma, with particular reference to their origin. Exp. Toxicol. Pathol. 2013, 65, 567–577. [Google Scholar] [CrossRef]
- Cai, X.; Lin, Y.; Friedrich, C.C.; Neville, C.; Pomerantseva, I.; Sundback, C.A.; Zhang, Z.; Vacanti, J.P.; Hauschka, P.V.; Grottkau, B.E. Bone marrow derived pluripotent cells are pericytes which contribute to vascularization. Stem Cell Rev. Rep. 2009, 5, 437–445. [Google Scholar] [CrossRef]
- Ebmeier, S.; Horsley, V. Origin of fibrosing cells in systemic sclerosis. Curr. Opin. Rheumatol. 2015, 27, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Marrelli, A.; Benedetto, P.D.; Liakouli, V.; Carubbi, F.; Ruscitti, P.; Alvaro, S.; Pantano, I.; Campese, A.F.; Grazioli, P.; et al. Scleroderma mesenchymal stem cells display a different phenotype from healthy controls; implications for regenerative medicine. Angiogenesis 2013, 16, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Guggino, G.; Zazzeroni, F.; Alesse, E.; et al. Perivascular cells in diffuse cutaneous systemic sclerosis overexpress activated ADAM12 and are involved in myofibroblast transdifferentiation and development of fibrosis. J. Rheumatol. 2016, 43, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Hegner, B.; Schaub, T.; Catar, R.; Kusch, A.; Wagner, P.; Essin, K.; Lange, C.; Riemekasten, G.; Dragun, D. Intrinsic deregulation of vascular smooth muscle and myofibroblast differentiation in mesenchymal stromal cells from patients with systemic sclerosis. PLoS ONE 2016, 11, e0153101. [Google Scholar] [CrossRef]
- Shi, J.; Yang, Y.; Cheng, A.; Xu, G.; He, F. Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H613–H631. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a reassessment. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front. Immunol. 2020, 11, 3053. [Google Scholar] [CrossRef] [PubMed]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Altorok, N.; Nada, S.; Kahaleh, B. The isolation and characterization of systemic sclerosis vascular smooth muscle cells: Enhanced proliferation and apoptosis resistance. J. Scleroderma Relat. Disord. 2016, 1, 307–311. [Google Scholar] [CrossRef]
- Svegliati, S.; Amico, D.; Spadoni, T.; Fischetti, C.; Finke, D.; Moroncini, G.; Paolini, C.; Tonnini, C.; Grieco, A.; Rovinelli, M.; et al. Agonistic anti-PDGF receptor autoantibodies from patients with systemic sclerosis impact human pulmonary artery smooth muscle cells function in vitro. Front. Immunol. 2017, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Tamama, K.; Sen, C.K.; Wells, A. Differentiation of bone marrow mesenchymal stem cells into the smooth muscle lineage by blocking ERK/MAPK signaling pathway. Stem Cells Dev. 2008, 17, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bendeck, M.P.; Simmons, C.A.; Santerre, J.P. Deriving vascular smooth muscle cells from mesenchymal stromal cells: Evolving differentiation strategies and current understanding of their mechanisms. Biomaterials 2017, 145, 9–22. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Hirai, S.; Tanaka, Y.; Sumi, T.; Tada, M.; Takahashi, H.; Watanabe, A.; Sakuma, Y. Pericyte-myofibroblast transition in the human lung. Biochem. Biophys. Res. Commun. 2020, 528, 269–275. [Google Scholar] [CrossRef]
- Wang, N.; Deng, Y.; Liu, A.; Shen, N.; Wang, W.; Du, X.; Tang, Q.; Li, S.; Odeh, Z.; Wu, T.; et al. Novel mechanism of the pericyte-myofibroblast transition in renal interstitial fibrosis: Core fucosylation regulation. Sci. Rep. 2017, 7, 16914. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chen, Q.; Luo, J.M.; Nie, J.; Meng, Q.H.; Shuai, W.; Xie, H.; Xia, J.M.; Wang, H. Notch1 promotes the pericyte-myofibroblast transition in idiopathic pulmonary fibrosis through the PDGFR/ROCK1 signal pathway. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Zent, J.; Guo, L.W. Signaling mechanisms of myofibroblastic activation: Outside-in and inside-out. Cell. Physiol. Biochem. 2018, 49, 848–868. [Google Scholar] [CrossRef]
- Lescoat, A.; Varga, J.; Matucci-Cerinic, M.; Khanna, D. New promising drugs for the treatment of systemic sclerosis: Pathogenic considerations, enhanced classifications, and personalized medicine. Expert Opin. Investig. Drugs 2021. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, C.; Allanore, Y. An update on targeted therapies in systemic sclerosis based on a systematic review from the last 3 years. Arthritis. Res. Ther. 2021, 23, 155. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gong, J.; Dennery, P.A.; Yao, H. Endothelial-to-mesenchymal transition: Pathogenesis and therapeutic targets for chronic pulmonary and vascular diseases. Biochem. Pharmacol. 2019, 168, 100–107. [Google Scholar] [CrossRef]
- Van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc pathophysiology; the myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Varga, J. Rationally-based therapeutic disease modification in systemic sclerosis: Novel strategies. Semin. Cell. Dev. Biol. 2020, 101, 146–160. [Google Scholar] [CrossRef]
- Hinchcliff, M.; O’Reilly, S. Current and potential new targets in systemic sclerosis therapy: A new hope. Curr. Rheumatol. Rep. 2020, 22, 42. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Sanchez Duffhues, G.; Ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 22, 3–13. [Google Scholar] [CrossRef]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Grembiale, R.D.; D’Antona, L.; Gallo, E.; D’Angelo, S.; Citraro, R.; Visca, P.; Olivieri, I.; De Sarro, G.; Perrotti, N.; et al. Oral metformin ameliorates bleomycin-induced skin fibrosis. J. Investig. Dermatol. 2016, 136, 1892–1894. [Google Scholar] [CrossRef]
- Mendoza, F.A.; Piera-Velazquez, S.; Jimenez, S.A. Tyrosine kinases in the pathogenesis of tissue fibrosis in systemic sclerosis and potential therapeutic role of their inhibition. Transl. Res. 2021, 231, 139–158. [Google Scholar] [CrossRef]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C. The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef]
- Huang, J.; Beyer, C.; Palumbo-Zerr, K.; Zhang, Y.; Ramming, A.; Distler, A.; Gelse, K.; Distler, O.; Schett, G.; Wollin, L.; et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann. Rheum. Dis. 2016, 75, 883–890. [Google Scholar] [CrossRef]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nagaoka, T.; Yoshida, T.; Wang, L.; Kuriyama, S.; Suzuki, Y.; Nagata, Y.; Harada, N.; Kodama, Y.; Takahashi, F.; et al. Nintedanib ameliorates experimental pulmonary arterial hypertension via inhibition of endothelial mesenchymal transition and smooth muscle cell proliferation. PLoS ONE 2019, 14, e0214697. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nintedanib: A review in fibrotic interstitial lung diseases. Drugs 2021, 81, 575–586. [Google Scholar] [CrossRef]
- Mura, M. Use of nintedanib in interstitial lung disease other than idiopathic pulmonary fibrosis: Much caution is warranted. Pulm. Pharmacol. Ther. 2021, 66, 101987. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Simultaneous inhibition of c-Abl and Src kinases abro-gates the exaggerated expression of profibrotic genes in cultured systemic sclerosis dermal fibroblasts. Clin. Exp. Rheumatol. 2018, 36, 36–44. [Google Scholar] [PubMed]
- Wermuth, P.J.; Jimenez, S.A. Abrogation of transforming growth factor-β-induced tissue fibrosis in TBRIcaCol1a2Cretransgenic mice by the second generation tyrosine kinase inhibitor SKI-606 (Bosutinib). PLoS ONE 2018, 13, e0196559. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jimenez, S.A. Protein kinase C delta and the c-abl kinase are required for transforming growth factor-beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011, 63, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Zhang, J.; Liu, T.; Shi, W. Rapamycin prevents endothelial cell migration by inhibiting the endothelial-to- mesenchymal transition and matrix metalloproteinase-2 and -9: An in vitro study. Mol. Vis. 2011, 17, 3406–3414. [Google Scholar]
- González-Mateo, G.T.; Aguirre, A.R.; Loureiro, J.; Abensur, H.; Sandoval, P.; Sánchez-Tomero, J.A.; del Peso, G.; Jiménez-Heffernan, J.A.; Ruiz-Carpio, V.; Selgas, R. Rapamycin protects from type-I peritoneal membrane failure inhibiting the angiogenesis, lymphangiogenesis, and Endo-MT. Biomed. Res. Int. 2015, 2015, 989560. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, A.; Yanaba, K.; Yoshizaki, A.; Iwata, Y.; Komura, K.; Ogawa, F.; Takenaka, M.; Shimizu, K.; Asano, Y.; Hasegawa, M. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin induced mouse models of systemic sclerosis. Arthritis Rheum. 2010, 62, 2476–2487. [Google Scholar] [CrossRef]
- Yoon, K.H. Proliferation signal inhibitors for the treatment of refractory autoimmune rheumatic diseases: A new therapeutic option. Ann. N. Y. Acad. Sci. 2009, 1173, 752–756. [Google Scholar] [CrossRef]
- Su, T.I.; Khanna, D.; Furst, D.E.; Danovitch, G.; Burger, C.; Maranian, P.; Clements, P.J. Rapamycin versus methotrexate in early diffuse systemic sclerosis: Results from a randomized, sin-gle-blind pilot study. Arthritis Rheum. 2009, 60, 3821–3830. [Google Scholar] [CrossRef]
- Fried, L.; Kirsner, R.S.; Bhandarkar, S.; Arbiser, J.L. Efficacy of rapamycin in scleroderma: A case study. Lymphat. Res. Biol. 2008, 6, 217–219. [Google Scholar] [CrossRef]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Rapamycin promotes endothelial-mesenchymal transition during stress-induced premature senescence through the activation of autophagy. Cell Commun. Signal. 2020, 18, 43. [Google Scholar] [CrossRef]
- Qi, Q.; Mao, Y.; Tian, Y.; Zhu, K.; Cha, X.; Wu, M.; Zhou, X. Geniposide inhibited endothelial–mesenchymal transition via the mTOR signaling pathway in a bleomycin-induced scleroderma mouse model. Am. J. Transl. Res. 2017, 15, 1025–1036. [Google Scholar]
- Jiang, Y.; Hu, F.; Li, Q.; Shen, C.; Yang, J.; Li, M. Tanshinone IIA ameliorates the bleomycin-induced endothelial-to-mesenchymal transition via the Akt/mTOR/p70S6K pathway in a murine model of systemic sclerosis. Int. Immunopharmacol. 2019, 77, 105968. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, J.; Li, M. Tanshinone IIA attenuates interleukin-17A-induced systemic sclerosis patient-derived dermal vascular smooth muscle cell activation via inhibition of the extracellular signal-regulated kinase signaling pathway. Clinics 2015, 70, 250–256. [Google Scholar] [CrossRef]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin β1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef]
- Soare, A.; Györfi, H.A.; Matei, A.E.; Dees, C.; Rauber, S.; Wohlfahrt, T.; Chen, C.W.; Ludolph, I.; Horch, R.E.; Bäuerle, T.; et al. Dipeptidylpeptidase 4 as a marker of activated fibroblasts and a potential target for the treatment of fibrosis in systemic sclerosis. Arthritis Rheumatol. 2020, 72, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Asano, Y.; Taniguchi, T.; Nakamura, K.; Saigusa, R.; Miura, S.; Toyama, T.; Takahashi, T.; Ichimura, Y.; Yoshizaki, A.; et al. Glycyrrhizin ameliorates fibrosis, vasculopathy, and inflammation in animal models of systemic sclerosis. J. Investig. Dermatol. 2017, 137, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Lakota, K.; Taniguchi, T.; Yoshizaki, A.; Sato, S.; Hong, W.; Zhou, X.; Sodin-Semrl, S.; Fang, F.; Asano, Y.; et al. An orally-active adiponectin receptor agonist mitigates cutaneous fibrosis, inflammation and microvascular pathology in a murine modelof systemic sclerosis. Sci. Rep. 2018, 8, 11843. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yang, F.; Hu, Y.; Chen, M.; Liu, Y.; Li, J.; Zhong, W. Hepatocyte growth factor attenuates the development of TGF-β1-induced EndMT through down-regulating the Notch signaling. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, J.; Zhou, X.; Chen, L.; Gong, Y.; Gao, Z.; Zhang, H.; Huang, W.; Zhou, H. Protective effect of spironolactone on endothelial-to-mesenchymal transition in HUVECs via Notch pathway. Cell Physiol. Biochem. 2015, 36, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Peppicelli, S.; Fabbrizzi, P.; Biagioni, A.; Mazzanti, B.; Menchi, G.; Calorini, L.; Pupi, A.; Trabocchi, A. Triazole RGD antagonist reverts TGFβ1-induced endothelial-to-mesenchymal transition in endothelial precursor cells. Mol. Cell. Biochem. 2017, 424, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Distler, J.H. Morphogen pathways in systemic sclerosis. Curr. Rheumatol. Rep. 2013, 15, 299. [Google Scholar] [CrossRef]
- Gyftaki-Venieri, D.A.; Abraham, D.J.; Ponticos, M. Insights into myofibroblasts and their activation in scleroderma: Op-portunities for therapy? Curr. Opin. Rheumatol. 2018, 30, 581–587. [Google Scholar] [CrossRef]
- Cisternas, P.; Vio, C.P.; Inestrosa, N.C. Role of Wnt signaling in tissue fibrosis, lessons from skeletal muscle and kidney. Curr. Mol. Med. 2014, 14, 510–522. [Google Scholar] [CrossRef]
- Wild, S.L.; Elghajiji, A.; Grimaldos Rodriguez, C.; Weston, S.D.; Burke, Z.D.; Tosh, D. The canonical Wnt pathway as a key regulator in liver development, differentiation and homeostatic renewal. Genes 2020, 11, 1163. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad dependent fibrotic responses in mesen-chymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef]
- Beyer, C.; Reichert, H.; Akan, H.; Mallano, T.; Schramm, A.; Dees, C.; Palumbo-Zerr, K.; Lin, N.Y.; Distler, A.; Gelse, K.; et al. Blockade of canonical Wnt signalling ameliorates experimental dermal fibrosis. Ann. Rheum. Dis. 2013, 72, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R.; Mantero, J.C.; Gordon, J.; Kishore, N.; Carns, M.; Dittrich, H.; Spiera, R.; Simms, R.W.; Varga, J. Inhibition of β-catenin signaling in the skin rescues cutaneous adipogenesis in systemic sclerosis: A randomized, double-blind, placebocon-trolled trial of C-82. J. Investig. Dermatol. 2017, 137, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Corciulo, C.; Liu, H.; Wilder, T.; Ito, M.; Cronstein, B. Adenosine A 2a receptor blockade diminishes Wnt/b-catenin signaling in a murine model of bleomycin-induced dermal fibrosis. Am. J. Pathol. 2017, 187, 1935–1944. [Google Scholar] [CrossRef]
- Bergmann, C.; Hallenberger, L.; Chenguiti Fakhouri, S.; Merlevede, B.; Brandt, A.; Dees, C.; Zhu, H.; Zehender, A.; Zhou, X.; Schwab, A.; et al. X-linked inhibitor of apoptosis protein (XIAP) inhibition in systemic sclerosis (SSc). Ann. Rheum. Dis. 2021. [Google Scholar] [CrossRef]
- Nandagopal, N.; Santat, L.A.; Elowitz, M.B. Cis-activation in the Notch signaling pathway. eLife 2019, 8, e37880. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, A.G.; El Hachem, M.; Zambruno, G.; Nystrom, A.; Candi, E.; Castiglia, D. Notch-ing up knowledge on molecular mechanisms of skin fibrosis: Focus on the multifaceted Notch signalling pathway. J. Biomed. Sci. 2021, 28, 36. [Google Scholar] [CrossRef]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Kavian, N.; Servettaz, A.; Mongaret, C.; Wang, A.; Nicco, C.; Chéreau, C.; Grange, P.; Vuiblet, V.; Birembaut, P.; Diebold, M.D.; et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheum. 2010, 62, 3477–3487. [Google Scholar] [CrossRef]
- Dees, C.; Zerr, P.; Tomcik, M.; Beyer, C.; Horn, A.; Akhmetshina, A.; Palumbo, K.; Reich, N.; Zwerina, J.; Sticherling, M.; et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum. 2011, 63, 1396–1404. [Google Scholar] [CrossRef]
- Tian, D.Y.; Jin, X.R.; Zeng, X.; Wang, Y. Notch signaling in endothelial cells: Is it the therapeutic target for vascular neointimal hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. [Google Scholar] [CrossRef]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell. 2011, 21, 288–300. [Google Scholar] [CrossRef]
- Liu, J.; Dong, F.; Jeong, J.; Masuda, T.; Lobe, C.G. Constitutively active Notch1 signaling promotes endothelial mesenchymal transition in a conditional transgenic mouse model. Int. J. Mol. Med. 2014, 34, 669–676. [Google Scholar] [CrossRef]
- Tian, D.; Zeng, X.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, Y. Protective effect of rapamycin on endothelial-to-mesenchymal transition in HUVECs through the Notch signaling pathway. Vascul. Pharmacol. 2019, 113, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.J.; Møller, L.B. Crosstalk of hedgehog and mTORC1 pathways. Cells 2020, 9, 2316. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Kireva, T.; Palumbo-Zerr, K.; Dees, C.; Tomcik, M.; Cordazzo, C.; Zerr, P.; Akhmetshina, A.; Ruat, M.; Distler, O.; et al. Inhibition of hedgehog signalling prevents experimental fibrosis and induces regression of established fibrosis. Ann. Rheum. Dis. 2012, 71, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.V.; Balani, P.; Lopez, A.R.; Nobleza, C.M.N.; Siddiqui, M.; Khan, S. A review of pirfenidone as an antifibrotic in idiopathic pulmonary fibrosis and its probable role in other diseases. Cureus 2021, 13, e12482. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, G.F.; Liao, X.P.; Li, X.J.; Zhang, J.; Lin, H.; Chen, Z.; Zhang, X. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int. J. Rheum. Dis. 2018, 21, 477–486. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.T.; Veijola, A.; Karvonen, H.; Lappi-Blanco, E.; Sormunen, R.; Korpela, S.; Zagai, U.; Sköld, M.C.; Kaarteenaho, R. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Albera, C.; Fischer, A.; Khalidi, N.; Raghu, G.; Chung, L.; Chen, D.; Schiopu, E.; Tagliaferri, M.; Seibold, J.R.; et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: The LOTUSS trial. J. Rheumatol. 2016, 43, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Distler, A.; Lang, V.; Del Vecchio, T.; Huang, J.; Zhang, Y.; Beyer, C.; Lin, N.Y.; Palumbo-Zerr, K.; Distler, O.; Schett, G.; et al. Combined inhibition of morphogen pathways demonstrates additive antifibrotic effects and improved tolerability. Ann. Rheum. Dis. 2014, 73, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Sun, M.; Sun, Y.; Feng, Z.; Kang, X.; Yang, W.; Wang, Y.; Luo, Y. New insights into the Hippo/YAP pathway in idiopathic pulmonary fibrosis. Pharmacol. Res. 2021, 169, 105635. [Google Scholar] [CrossRef]
- Toyama, T.; Looney, A.P.; Baker, B.M.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J. Investig. Dermatol. 2018, 138, 78–88. [Google Scholar] [CrossRef]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. J. Scleroderma Relat. Disord. 2020, 5, 40–50. [Google Scholar] [CrossRef]
- You, H.; Xu, D.; Zhao, J.; Li, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. JAK inhibitors: Prospects in connective tissue diseases. Clin. Rev. Allergy Immunol. 2020, 59, 334–351. [Google Scholar] [CrossRef]
- Roger, I.; Milara, J.; Montero, P.; Cortijo, J. The role of JAK/STAT molecular pathway in vascular remodeling associated with pulmonary hypertension. Int. J. Mol. Sci. 2021, 22, 4980. [Google Scholar] [CrossRef]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef]
- Pedroza, M.V.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast myofibroblast differentiation. FASEB J. 2016, 30, 129–140. [Google Scholar] [CrossRef]
- Dees, C.; Tomcik, M.; Palumbo-Zerr, K.; Distler, A.; Beyer, C.; Lang, V.; Horn, A.; Zerr, P.; Zwerina, J.; Gelse, K.; et al. JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 2012, 64, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis post transcriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell. 2018, 29, 84–95. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef]
- Jing, J.; Dou, T.T.; Yang, J.Q.; Chen, X.B.; Cao, H.L.; Min, M.; Cai, S.Q.; Zheng, M.; Man, X.Y. Role of endothelin-1 in the skin fibrosis of systemic sclerosis. Eur. Cytokine Netw. 2015, 26, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, P.J.; Li, Z.; Mendoza, F.A.; Jimenez, S.A. Stimulation of transforming growth factor- β1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): A novel profibrotic effect of ET-1. PLoS ONE 2016, 11, e0161988. [Google Scholar] [CrossRef] [PubMed]
- Soldano, S.; Paolino, S.; Pizzorni, C.; Trombetta, A.C.; Montagna, P.; Brizzolara, R.; Corallo, C.; Giordano, N.; Sulli, A.; Cutolo, M. Dual endothelin receptor antagonists contrast the effects induced by endothelin-1 on cultured human microvascular endothelial cells. Clin. Exp. Rheumatol. 2017, 35, 484–493. [Google Scholar] [PubMed]
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res. Ther. 2017, 19, 134. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Tsou, P.S.; Amin, M.A.; Ruth, J.H.; Campbell, P.; Fox, D.A.; Khanna, D.; Larsen, S.D.; Neubig, R.R. Targeting the myofibroblast genetic switch: Inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J. Pharmacol. Exp. Ther. 2014, 349, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Kahl, D.J.; Hutchings, K.M.; Lisabeth, E.M.; Haak, A.J.; Leipprandt, J.R.; Dexheimer, T.; Khanna, D.; Tsou, P.S.; Campbell, P.L.; Fox, D.A.; et al. 5-Aryl-1,3,4-oxadiazol-2-ylthioalkanoic acids: A highly potent new class of inhibitors of rho/myocardin-related transcription factor (mrtf)/serum response factor (srf)-mediated gene transcription as potential antifibrotic agents for scleroderma. J. Med. Chem. 2019, 62, 4350–4369. [Google Scholar] [CrossRef]
- Hutchings, K.M.; Lisabeth, E.M.; Rajeswaran, W.; Wilson, M.W.; Sorenson, R.J.; Campbell, P.L.; Ruth, J.H.; Amin, A.; Tsou, P.S.; Leipprandt, J.R.; et al. Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorg. Med. Chem. Lett. 2017, 27, 1744–1749. [Google Scholar] [CrossRef]
- Denton, C.P.; Ong, V.H.; Xu, S.; Chen-Harris, H.; Modrusan, Z.; Lafyatis, R.; Khanna, D.; Jahreis, A.; Siegel, J.; Sornasse, T. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: Insights from the faSScinate clinical trial in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1362–1371. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Lin, C.J.F.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: Results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis. 2018, 77, 212–220. [Google Scholar] [CrossRef]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Mantero, J.C.; Kishore, N.; Ziemek, J.; Stifano, G.; Zammitti, C.; Khanna, D.; Gordon, J.K.; Spiera, R.; Zhang, Y.; Simms, R.W.; et al. Randomised, double-blind, placebo-controlled trial of IL1-trap, rilonacept, in systemic sclerosis. A phase I/II biomarker trial. Clin. Exp. Rheumatol. 2018, 36, 146–149. [Google Scholar]
- Mor, A.; Segal Salto, M.; Katav, A.; Barashi, N.; Edelshtein, V.; Manetti, M.; Levi, Y.; George, J.; Matucci-Cerinic, M. Blockade of CCL24 with a monoclonal antibody ameliorates experimental dermal and pulmonary fibrosis. Ann. Rheum. Dis. 2019, 78, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Konstantinova, I.; Guignabert, C.; Pezet, S.; Sadoine, J.; Guilbert, T.; Cauvet, A.; Tu, L.; Luccarini, J.M.; Junien, J.L.; et al. Pan-PPAR agonist IVA337 is effective in experimental lung fibrosis and pulmonary hypertension. Ann. Rheum. Dis. 2017, 76, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Ruzehaji, N.; Frantz, C.; Ponsoye, M.; Avouac, J.; Pezet, S.; Guilbert, T.; Luccarini, J.M.; Broqua, P.; Junien, J.L.; Allanore, Y. Pan PPAR agonist IVA337 is effective in prevention and treatment of experimental skin fibrosis. Ann. Rheum. Dis. 2016, 75, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Asano, Y.; Akamata, K.; Noda, S.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Shudo, K.; Sato, S.; Kadono, T. Tamibarotene ameliorates bleomycin-induced dermal fibrosis by modulating phenotypes of fibroblasts, endothelial cells, and immune cells. J. Investig. Dermatol. 2016, 136, 387–398. [Google Scholar] [CrossRef]
- Tsou, P.S.; Palisoc, P.J.; Flavahan, N.A.; Khanna, D. Dissecting the cellular mechanism of prostacyclin analogue iloprost in reversing vascular dysfunction in scleroderma. Arthritis Rheumatol. 2020, 73, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Peng, L.; Liu, P.; Yang, M.; Zhou, H.; Ding, Y.; Wang, J.; Huang, W.; Tan, Q.; Wang, Y.; et al. Paeoniflorin ameliorates chronic Hypoxia/SU5416-induced pulmonary arterial hypertension by inhibiting endothelial-to-mesenchymal transition. Drug Des. Devel. Ther. 2020, 14, 1191–1202. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef]
- Khanna, D.; Allanore, Y.; Denton, C.P.; Kuwana, M.; Matucci-Cerinic, M.; Pope, J.E.; Atsumi, T.; Bečvář, R.; Czirják, L.; Hachulla, E.; et al. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): Randomised, double-blind, placebo-controlled multicentre trial. Ann. Rheum. Dis. 2020, 79, 618–625. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, E.; Selvi, E.; Balistreri, E.; Lorenzini, S.; Maggio, R.; Natale, M.R.; Capecchi, P.L.; Lazzerini, P.E.; Bardelli, M.; Laghi-Pasini, F.; et al. Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology 2009, 48, 1050–1056. [Google Scholar] [CrossRef]
- Balistreri, E.; Garcia-Gonzalez, E.; Selvi, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Maggio, R.; Lucattelli, M.; Galeazzi, M.; Distler, J.W. The cannabinoid WIN55, 212-2 abrogates dermal fibrosis in scleroderma bleomycin model. Ann. Rheum. Dis. 2011, 70, 695–699. [Google Scholar] [CrossRef]
- Fu, Q.; Zheng, Y.; Dong, X.; Wang, L.; Jiang, C.G. Activation of cannabinoid receptor type 2 by JWH133 alleviates bleomycin-induced pulmonary fibrosis in mice. Oncotarget 2017, 8, 103486–1034898. [Google Scholar] [CrossRef]
- Spiera, R.; Hummers, L.; Chung, L.; Frech, T.M.; Domsic, R.; Hsu, V.; Furst, D.E.; Gordon, J.; Mayes, M.; Simms, R.; et al. Safety and efficacy of lenabasum in a phase II, randomized, placebo-controlled trial in adults with systemic sclerosis. Arthritis Rheumatol. 2020, 72, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Liakouli, V.; Ruscitti, P.; Berardicurti, O.; Carubbi, F.; Panzera, N.; Di Bartolomeo, S.; Guggino, G.; Ciccia, F.; Triolo, G.; et al. Blocking CD248 molecules in perivascular stromal cells of patients with systemic sclerosis strongly inhibits their differentiation toward myofibroblasts and proliferation: A new potential target for antifibrotic therapy. Arthritis Res. Ther. 2018, 20, 223. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, E.; Rosa, I.; Fioretto, B.S.; Matucci-Cerinic, M.; Manetti, M. New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life 2021, 11, 610. https://doi.org/10.3390/life11070610
Romano E, Rosa I, Fioretto BS, Matucci-Cerinic M, Manetti M. New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life. 2021; 11(7):610. https://doi.org/10.3390/life11070610
Chicago/Turabian StyleRomano, Eloisa, Irene Rosa, Bianca Saveria Fioretto, Marco Matucci-Cerinic, and Mirko Manetti. 2021. "New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy" Life 11, no. 7: 610. https://doi.org/10.3390/life11070610
APA StyleRomano, E., Rosa, I., Fioretto, B. S., Matucci-Cerinic, M., & Manetti, M. (2021). New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life, 11(7), 610. https://doi.org/10.3390/life11070610