
Effects of CNS Injury-Induced Immunosuppression on Pulmonary Immunity




Abstract
1. Introduction
2. Community-Acquired Pneumonia
3. Ventilator-Acquired Pneumonia
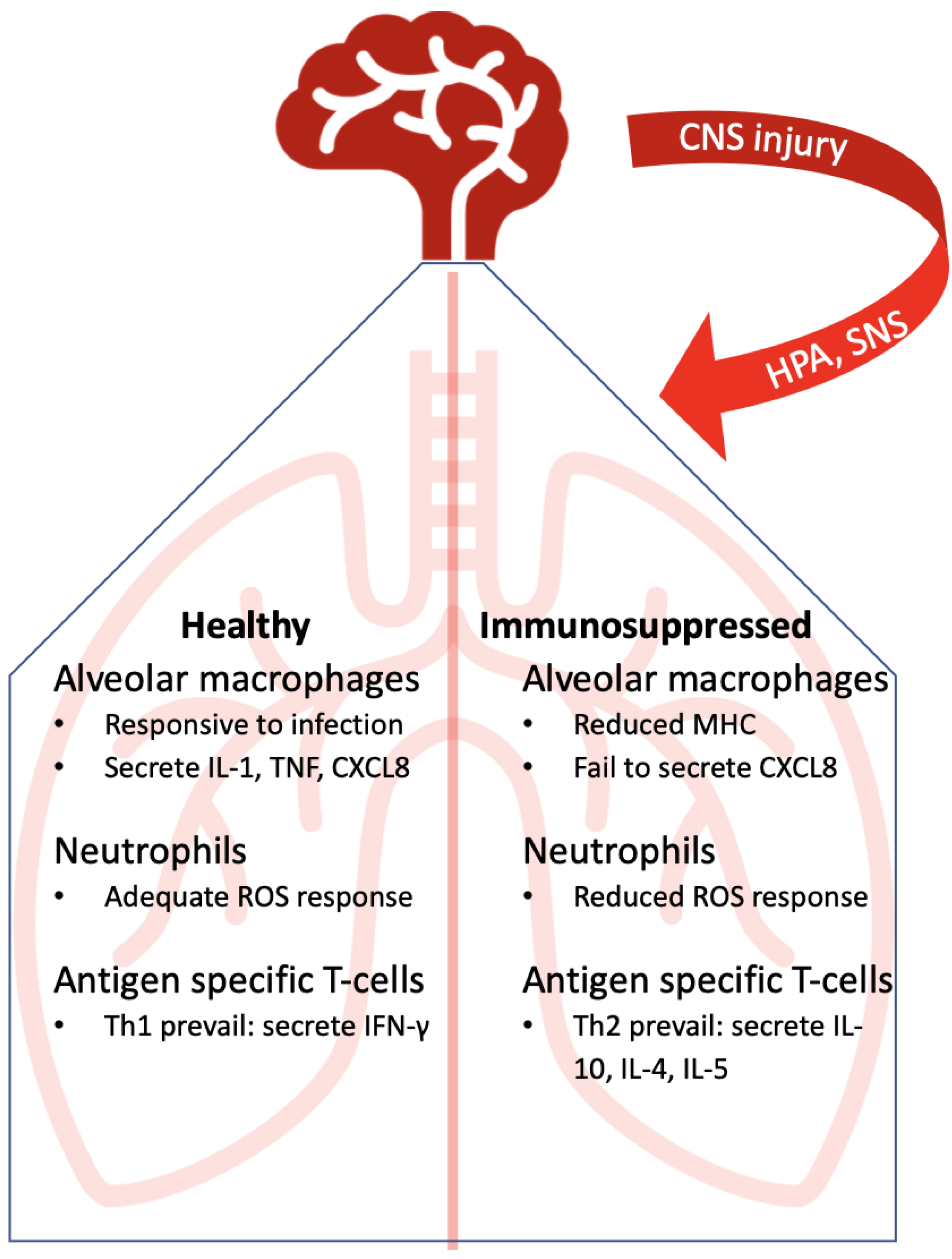
4. Immunocompetent Lung Immunity Against Lower Respiratory Tract Infections
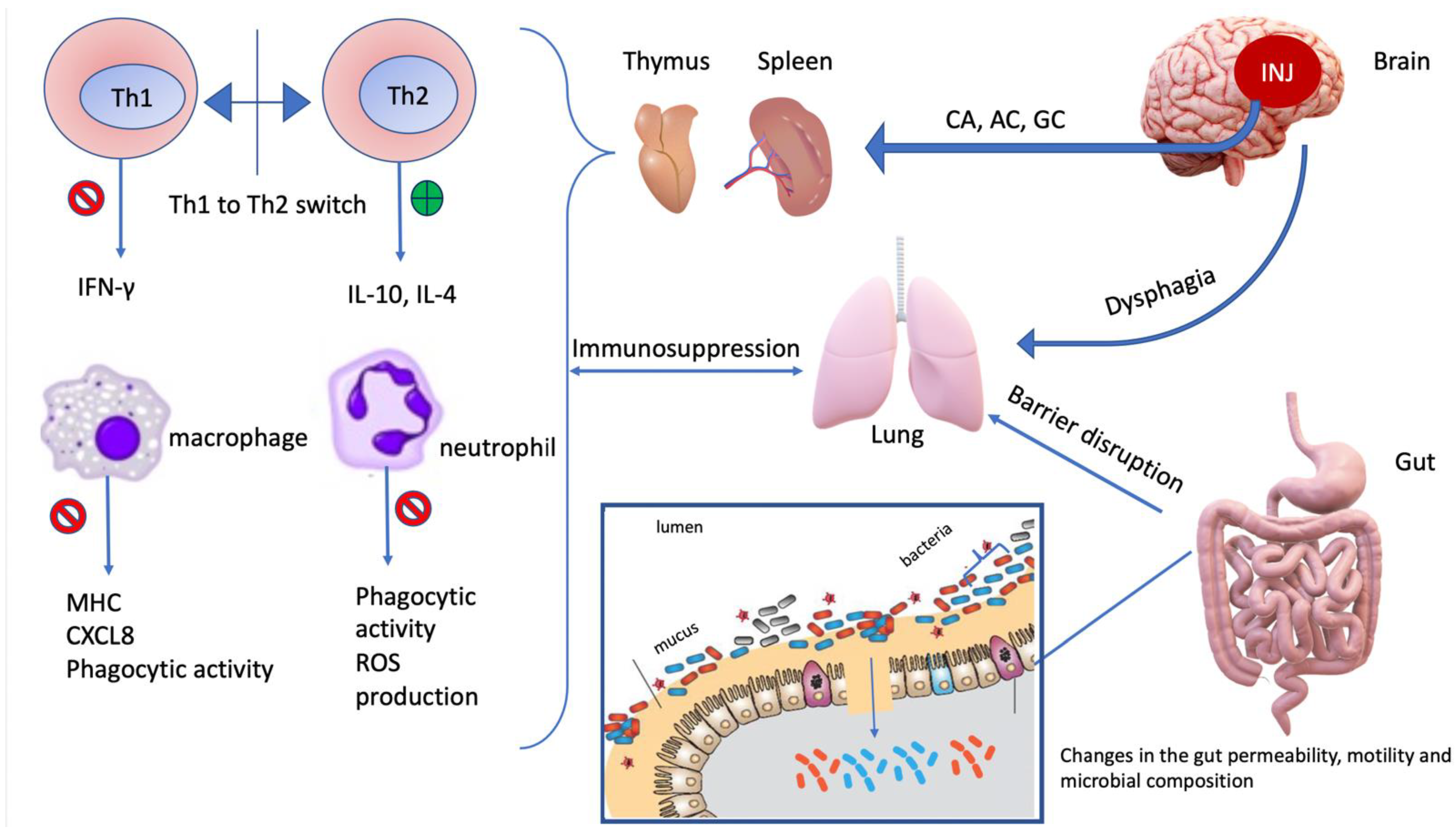
5. Immunosuppressed Lung: Points of Disruption
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic Cascades in Ischemic Stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- García-Berrocoso, T.; Giralt, D.; Llombart, V.; Bustamante, A.; Penalba, A.; Flores, A.; Ribó, M.; Molina, C.A.; Rosell, A.; Montaner, J. Chemokines after Human Ischemic Stroke: From Neurovascular Unit to Blood Using Protein Arrays. Transl. Proteom. 2014, 3, 1–9. [Google Scholar] [CrossRef]
- Catania, A.; Lonati, C.; Sordi, A.; Gatti, S. Detrimental Consequences of Brain Injury on Peripheral Cells. Brain. Behav. Immun. 2009, 23, 877–884. [Google Scholar] [CrossRef]
- Bobbo, V.C.D.; Jara, C.P.; Mendes, N.F.; Morari, J.; Velloso, L.A.; Araújo, E.P. Interleukin-6 Expression by Hypothalamic Microglia in Multiple Inflammatory Contexts: A Systematic Review. BioMed Res. Int. 2019, 2019, 1365210. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-J.; Hsu, C.-C.; Chio, C.-C.; Tian, Y.-F.; Lin, M.-T.; Lin, T.-W.; Chang, C.-H.; Chang, C.-P. Gamma-Secretase Inhibitors Attenuate Neurotrauma and Neurogenic Acute Lung Injury in Rats by Rescuing the Accumulation of Hypertrophic Microglia. Cell. Physiol. Biochem. 2017, 44, 1726–1740. [Google Scholar] [CrossRef]
- Zhou, J.; Noori, H.; Burkovskiy, I.; Lafreniere, J.; Kelly, M.; Lehmann, C. Modulation of the Endocannabinoid System Following Central Nervous System Injury. Int. J. Mol. Sci. 2019, 20, 388. [Google Scholar] [CrossRef] [PubMed]
- Westendorp, W.F.; Nederkoorn, P.J.; Vermeij, J.-D.; Dijkgraaf, M.G.; van de Beek, D. Post-Stroke Infection: A Systematic Review and Meta-Analysis. BMC Neurol. 2011, 11, 110. [Google Scholar] [CrossRef]
- Finlayson, O.; Kapral, M.; Hall, R.; Asllani, E.; Selchen, D.; Saposnik, G.; Canadian Stroke Network; Stroke Outcome Research Canada (SORCan) Working Group. Risk Factors, Inpatient Care, and Outcomes of Pneumonia after Ischemic Stroke. Neurology 2011, 77, 1338–1345. [Google Scholar] [CrossRef]
- Ramirez, J.A.; Wiemken, T.L.; Peyrani, P.; Arnold, F.W.; Kelley, R.; Mattingly, W.A.; Nakamatsu, R.; Pena, S.; Guinn, B.E.; Furmanek, S.P.; et al. Adults Hospitalized With Pneumonia in the United States: Incidence, Epidemiology, and Mortality. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017, 65, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Kishore, A.K.; Vail, A.; Chamorro, A.; Garau, J.; Hopkins, S.J.; Di Napoli, M.; Kalra, L.; Langhorne, P.; Montaner, J.; et al. Diagnosis of Stroke-Associated Pneumonia: Recommendations From the Pneumonia in Stroke Consensus Group. Stroke 2015, 46, 2335–2340. [Google Scholar] [CrossRef] [PubMed]
- Frankel, H.L.; Coll, J.R.; Charlifue, S.W.; Whiteneck, G.G.; Gardner, B.P.; Jamous, M.A.; Krishnan, K.R.; Nuseibeh, I.; Savic, G.; Sett, P. Long-Term Survival in Spinal Cord Injury: A Fifty Year Investigation. Spinal Cord 1998. [Google Scholar] [CrossRef]
- Woratyla, S.P.; Morgan, A.S.; Mackay, L.; Bernstein, B.; Barba, C. Factors Associated with Early Onset Pneumonia in the Severely Brain-Injured Patient. Conn. Med. 1995, 59, 643–647. [Google Scholar]
- Evans, C.; Weaver, F.; Rogers, T.; Rapacki, L.; Miskevics, S.; Hahm, B.; Smith, B.; Lavela, S.; Goldstein, B.; Burns, S. Guideline-Recommended Management of Community-Acquired Pneumonia in Veterans with Spinal Cord Injury. Top. Spinal Cord Inj. Rehabil. 2012. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Roig, I.L.; Cazares, G.; Stager, C.E.; Logan, N.; Safar, H. Can an Etiologic Agent Be Identified in Adults Who Are Hospitalized for Community-Acquired Pneumonia: Results of a One-Year Study. J. Infect. 2013, 67, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Strålin, K.; Olcén, P.; Törnqvist, E.; Holmberg, H. Definite, Probable, and Possible Bacterial Aetiologies of Community-Acquired Pneumonia at Different CRB-65 Scores. Scand. J. Infect. Dis. 2010, 42, 426–434. [Google Scholar] [CrossRef]
- Arancibia, F.; Bauer, T.T.; Ewig, S.; Mensa, J.; Gonzalez, J.; Niederman, M.S.; Torres, A. Community-Acquired Pneumonia Due to Gram-Negative Bacteria and Pseudomonas Aeruginosa: Incidence, Risk, and Prognosis. Arch. Intern. Med. 2002. [Google Scholar] [CrossRef]
- Fuller, G.W.; Ransom, J.; Mandrekar, J.; Brown, A.W. Long-Term Survival Following Traumatic Brain Injury: A Population-Based Parametric Survival Analysis. Neuroepidemiology 2016. [Google Scholar] [CrossRef]
- Torres, A.; Peetermans, W.E.; Viegi, G.; Blasi, F. Risk Factors for Community-Acquired Pneumonia in Adults in Europe: A Literature Review. Thorax 2013. [Google Scholar] [CrossRef]
- Koenig, S.M.; Truwit, J.D. Ventilator-Associated Pneumonia: Diagnosis, Treatment, and Prevention. Clin. Microbiol. Rev. 2006, 19, 637–657. [Google Scholar] [CrossRef]
- Hui, X.; Haider, A.H.; Hashmi, Z.G.; Rushing, A.P.; Dhiman, N.; Scott, V.K.; Selvarajah, S.; Haut, E.R.; Efron, D.T.; Schneider, E.B. Increased Risk of Pneumonia among Ventilated Patients with Traumatic Brain Injury: Every Day Counts! J. Surg. Res. 2013, 184, 438–443. [Google Scholar] [CrossRef]
- Hu, P.J.; Pittet, J.-F.; Kerby, J.D.; Bosarge, P.L.; Wagener, B.M. Acute Brain Trauma, Lung Injury, and Pneumonia: More than Just Altered Mental Status and Decreased Airway Protection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1–L15. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, M.I.; Peterson, J.; Fernandez, J.F.; Qin, Z.; Fisher, A.C.; Nicholson, S.C. Comparison of the Bacterial Etiology of Early-Onset and Late-Onset Ventilator-Associated Pneumonia in Subjects Enrolled in 2 Large Clinical Studies. Respir. Care 2013, 58, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Giantsou, E.; Liratzopoulos, N.; Efraimidou, E.; Panopoulou, M.; Alepopoulou, E.; Kartali-Ktenidou, S.; Minopoulos, G.I.; Zakynthinos, S.; Manolas, K.I. Both Early-Onset and Late-Onset Ventilator-Associated Pneumonia Are Caused Mainly by Potentially Multiresistant Bacteria. Intensive Care Med. 2005, 31, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, Y.; Hargett, J.L.; Lenhardt, R.; Heine, M.F.; Doufas, A.G.; Remmel, K.S.; Ramirez, J.A.; Akça, O. Ventilator-Associated Pneumonia in Critically Ill Stroke Patients: Frequency, Risk Factors, and Outcomes. J. Crit. Care 2011, 26, 273–279. [Google Scholar] [CrossRef]
- Jovanovic, B.; Milan, Z.; Markovic-Denic, L.; Djuric, O.; Radinovic, K.; Doklestic, K.; Velickovic, J.; Ivancevic, N.; Gregoric, P.; Pandurovic, M.; et al. Risk Factors for Ventilator-Associated Pneumonia in Patients with Severe Traumatic Brain Injury in a Serbian Trauma Centre. Int. J. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Eddens, T.; Kolls, J.K. Host Defenses against Bacterial Lower Respiratory Tract Infection. Curr. Opin. Immunol. 2012, 24, 424–430. [Google Scholar] [CrossRef]
- Zhang, P.; Summer, W.R.; Bagby, G.J.; Nelson, S. Innate Immunity and Pulmonary Host Defense: Lung Innate Immunity. Immunol. Rev. 2000, 173, 39–51. [Google Scholar] [CrossRef]
- Hussell, T.; Bell, T.J. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Aberdein, J.D.; Cole, J.; Bewley, M.A.; Marriott, H.M.; Dockrell, D.H. Alveolar Macrophages in Pulmonary Host Defence-the Unrecognized Role of Apoptosis as a Mechanism of Intracellular Bacterial Killing. Clin. Exp. Immunol. 2013, 174, 193–202. [Google Scholar]
- Mizgerd, J.P. Mechanisms of Disease: Acute Lower Respiratory Tract Infection. N. Engl. J. Med. 2008, 358, 716–727. [Google Scholar] [CrossRef]
- Grommes, J.; Soehnlein, O. Contribution of Neutrophils to Acute Lung Injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef]
- Mizgerd, J.P.; Spieker, M.R.; Doerschuk, C.M. Early Response Cytokines and Innate Immunity: Essential Roles for TNF Receptor 1 and Type I IL-1 Receptor During Escherichia Coli Pneumonia in Mice. J. Immunol. 2001, 166, 4042–4048. [Google Scholar] [CrossRef]
- Bullard, D.C.; Kunkel, E.J.; Kubo, H.; Hicks, M.J.; Lorenzo, I.; Doyle, N.A.; Doerschuk, C.M.; Ley, K.; Beaudet, A.L. Infectious Susceptibility and Severe Deficiency of Leukocyte Rolling and Recruitment in E-Selectin and P-Selectin Double Mutant Mice. J. Exp. Med. 1996, 183, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Quinlan, W.M.; Doyle, N.A.; Graham, L.; Sligh, J.E.; Takei, F.; Beaudet, A.L.; Doerschuk, C.M. The Roles of CD11/CD18 and ICAM-1 in Acute Pseudomonas Aeruginosa-Induced Pneumonia in Mice. J. Immunol. 1996, 157, 5016–5021. [Google Scholar] [PubMed]
- Shimizu, T.; Kida, Y.; Kuwano, K. A Dipalmitoylated Lipoprotein from Mycoplasma Pneumoniae Activates NF-ΚB through TLR1, TLR2, and TLR6. J. Immunol. 2005, 175, 4641–4646. [Google Scholar] [CrossRef]
- Cao, F.; Castrillo, A.; Tontonoz, P.; Re, F.; Byrne, G.I. Chlamydia Pneumoniae-Induced Macrophage Foam Cell Formation Is Mediated by Toll-like Receptor 2. Infect. Immun. 2007, 75, 753–759. [Google Scholar] [CrossRef]
- Tudhope, S.J.; Catley, M.C.; Fenwick, P.S.; Russell, R.E.K.; Rumsey, W.L.; Newton, R.; Barnes, P.J.; Donnelly, L.E. The Role of IκB Kinase 2, but Not Activation of NF-ΚB, in the Release of CXCR3 Ligands from IFN-γ-Stimulated Human Bronchial Epithelial Cells. J. Immunol. 2007, 179, 6237–6245. [Google Scholar] [CrossRef]
- Gunasekera, P.; Gratrix, A. Ventilator-Associated Pneumonia. BJA Educ. 2016, 16, 198–202. [Google Scholar] [CrossRef]
- Pennington, J.E.; Ehrie, M.G. Pathogenesis of Pseudomonas Aeruginosa Pneumonia during Immunosuppression. J. Infect. Dis. 1978, 137, 764–774. [Google Scholar] [CrossRef]
- Brommer, B.; Engel, O.; Kopp, M.A.; Watzlawick, R.; Müller, S.; Prüss, H.; Chen, Y.; DeVivo, M.J.; Finkenstaedt, F.W.; Dirnagl, U.; et al. Spinal Cord Injury-Induced Immune Deficiency Syndrome Enhances Infection Susceptibility Dependent on Lesion Level. Brain 2016, 139, 692–707. [Google Scholar] [CrossRef] [PubMed]
- Shim, R.; Wong, C.H.Y. Complex Interplay of Multiple Biological Systems That Contribute to Post-Stroke Infections. Brain. Behav. Immun. 2018. [Google Scholar] [CrossRef]
- Doran, S.J.; Henry, R.J.; Shirey, K.A.; Barrett, J.P.; Ritzel, R.M.; Lai, W.; Blanco, J.C.; Faden, A.I.; Vogel, S.N.; Loane, D.J. Early or Late Bacterial Lung Infection Increases Mortality After Traumatic Brain Injury in Male Mice and Chronically Impairs Monocyte Innate Immune Function. Crit. Care Med. 2020, 48, e418–e428. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.W.; Yang, H.; Yan, F.L.; Wang, H.; Xing, F.L.; Zuo, L.; Zhang, H.Q. Blocking Sympathetic Nervous System Reverses Partially Stroke-Induced Immunosuppression but Does Not Aggravate Functional Outcome After Experimental Stroke in Rats. Neurochem. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wolach, B.; Sazbon, L.; Gavrieli, R.; Broda, A.; Schlesinger, M. Early Immunological Defects in Comatose Patients after Acute Brain Injury. J. Neurosurg. 2001, 94, 706–711. [Google Scholar] [CrossRef]
- Draxler, D.F.; Awad, M.M.; Hanafi, G.; Daglas, M.; Ho, H.; Keragala, C.; Galle, A.; Roquilly, A.; Lyras, D.; Sashindranath, M.; et al. Tranexamic Acid Influences the Immune Response, but Not Bacterial Clearance in a Model of Post-Traumatic Brain Injury Pneumonia. J. Neurotrauma 2019. [Google Scholar] [CrossRef]
- Draxler, D.F.; Daglas, M.; Fernando, A.; Hanafi, G.; McCutcheon, F.; Ho, H.; Galle, A.; Gregory, J.; Larsson, P.; Keragala, C.; et al. Tranexamic Acid Modulates the Cellular Immune Profile after Traumatic Brain Injury in Mice without Hyperfibrinolysis. J. Thromb. Haemost. 2019, 17, 2174–2187. [Google Scholar] [CrossRef]
- Muehlstedt, S.G.; Lyte, M.; Rodriguez, J.L. Increased IL-10 Production and HLA-DR Suppression in the Lungs of Injured Patients Precede the Development of Nosocomial Pneumonia. Shock 2002, 17. [Google Scholar] [CrossRef]
- Muehlstedt, S.G.; Richardson, C.J.; West, M.A.; Lyte, M.; Rodriguez, J.L. Cytokines and the Pathogenesis of Nosocomial Pneumonia. Surgery 2001, 130, 602–611. [Google Scholar] [CrossRef]
- Członkowska, A.; Cyrta, B.; Korlak, J. Immunological Observations on Patients with Acute Cerebral Vascular Disease. J. Neurol. Sci. 1979, 43, 455–464. [Google Scholar] [CrossRef]
- Haeusler, K.G.; Schmidt, W.U.H.; Föhring, F.; Meisel, C.; Helms, T.; Jungehulsing, G.J.; Nolte, C.H.; Schmolke, K.; Wegner, B.; Meisel, A.; et al. Cellular Immunodepression Preceding Infectious Complications after Acute Ischemic Stroke in Humans. Cerebrovasc. Dis. 2008, 25, 50–58. [Google Scholar] [CrossRef]
- Prass, K.; Meisel, C.; Höflich, C.; Braun, J.; Halle, E.; Wolf, T.; Ruscher, K.; Victorov, I.V.; Priller, J.; Dirnagl, U.; et al. Stroke-Induced Immunodeficiency Promotes Spontaneous Bacterial Infections and Is Mediated by Sympathetic Activation Reversal by Poststroke T Helper Cell Type 1-like Immunostimulation. J. Exp. Med. 2003, 198, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Farris, B.Y.; Monaghan, K.L.; Zheng, W.; Amend, C.D.; Hu, H.; Ammer, A.G.; Coad, J.E.; Ren, X.; Wan, E.C.K. Ischemic Stroke Alters Immune Cell Niche and Chemokine Profile in Mice Independent of Spontaneous Bacterial Infection. Immun. Inflamm. Dis. 2019, 7, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Mracsko, E.; Liesz, A.; Karcher, S.; Zorn, M.; Bari, F.; Veltkamp, R. Differential Effects of Sympathetic Nervous System and Hypothalamic-Pituitary-Adrenal Axis on Systemic Immune Cells after Severe Experimental Stroke. Brain. Behav. Immun. 2014, 41, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Römer, C.; Engel, O.; Winek, K.; Hochmeister, S.; Zhang, T.; Royl, G.; Klehmet, J.; Dirnagl, U.; Meisel, C.; Meisel, A. Blocking Stroke-Induced Immunodeficiency Increases CNS Antigen-Specific Autoreactivity but Does Not Worsen Functional Outcome after Experimental Stroke. J. Neurosci. 2015, 35, 7777–7794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, F.; Leak, R.K.; Chen, J.; Hu, X. Regulation of Neuroinflammation through Programed Death-1/Programed Death Ligand Signaling in Neurological Disorders. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Moon, Y.-H.; Hyung, K.E.; Yoo, J.-S.; Lee, M.J.; Lee, I.H.; Go, B.S.; Hwang, K.W. Macrophage PD-L1 Strikes Back: PD-1/PD-L1 Interaction Drives Macrophages toward Regulatory Subsets. Adv. Biosci. Biotechnol. 2013, 4, 19–29. [Google Scholar] [CrossRef]
- Yamashiro, K.; Tanaka, R.; Urabe, T.; Ueno, Y.; Yamashiro, Y.; Nomoto, K.; Takahashi, T.; Tsuji, H.; Asahara, T.; Hattori, N. Gut Dysbiosis Is Associated with Metabolism and Systemic Inflammation in Patients with Ischemic Stroke. PLoS ONE 2017, 12, e0171521. [Google Scholar] [CrossRef]
- Stanley, D.; Moore, R.J.; Wong, C.H.Y. An Insight into Intestinal Mucosal Microbiota Disruption after Stroke. Sci. Rep. 2018, 8, 568. [Google Scholar] [CrossRef]
- Wang, L.; He, Y.; Li, H.; Ai, Q.; Yu, J. The Microbiota Protects against Pseudomonas Aeruginosa Pneumonia via Γδ T Cell-Neutrophil Axis in Mice. Microbes Infect. 2020, 22, 294–302. [Google Scholar] [CrossRef]
- McKenzie, D.R.; Kara, E.E.; Bastow, C.R.; Tyllis, T.S.; Fenix, K.A.; Gregor, C.E.; Wilson, J.J.; Babb, R.; Paton, J.C.; Kallies, A.; et al. IL-17-Producing Γδ T Cells Switch Migratory Patterns between Resting and Activated States. Nat. Commun. 2017, 8, 15632. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Readout | Outcome |
|---|---|---|
| Macrophages | Recruitment | ↑ |
| Antigen presentation | ↓ | |
| Respiratory burst | ↓ | |
| Pro-inflammatory cytokines | ↓ | |
| Neutrophils | Recruitment | ↑ |
| Respiratory burst | ↓ | |
| Phagocytosis | ↓ | |
| T-cells | Counts | ↓ |
| IFN-gamma | ↓ | |
| Th1 | ↓ | |
| Il-4 | ↑ | |
| Th2 | ↑ | |
| Pro-inflammatory cytokines | ↓ | |
| Apoptosis | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bietar, B.; Lehmann, C.; Stadnyk, A.W. Effects of CNS Injury-Induced Immunosuppression on Pulmonary Immunity. Life 2021, 11, 576. https://doi.org/10.3390/life11060576
Bietar B, Lehmann C, Stadnyk AW. Effects of CNS Injury-Induced Immunosuppression on Pulmonary Immunity. Life. 2021; 11(6):576. https://doi.org/10.3390/life11060576
Chicago/Turabian StyleBietar, Bashir, Christian Lehmann, and Andrew W. Stadnyk. 2021. "Effects of CNS Injury-Induced Immunosuppression on Pulmonary Immunity" Life 11, no. 6: 576. https://doi.org/10.3390/life11060576
APA StyleBietar, B., Lehmann, C., & Stadnyk, A. W. (2021). Effects of CNS Injury-Induced Immunosuppression on Pulmonary Immunity. Life, 11(6), 576. https://doi.org/10.3390/life11060576