
Altered Bone Status in Rett Syndrome

 , ,
, , 


Abstract
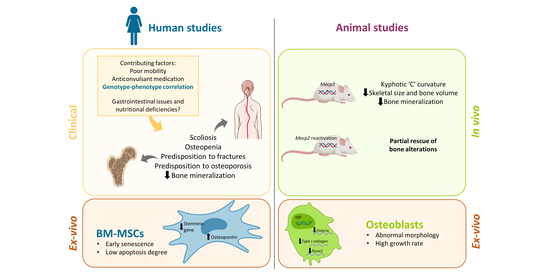
1. Introduction: Rett Syndrome
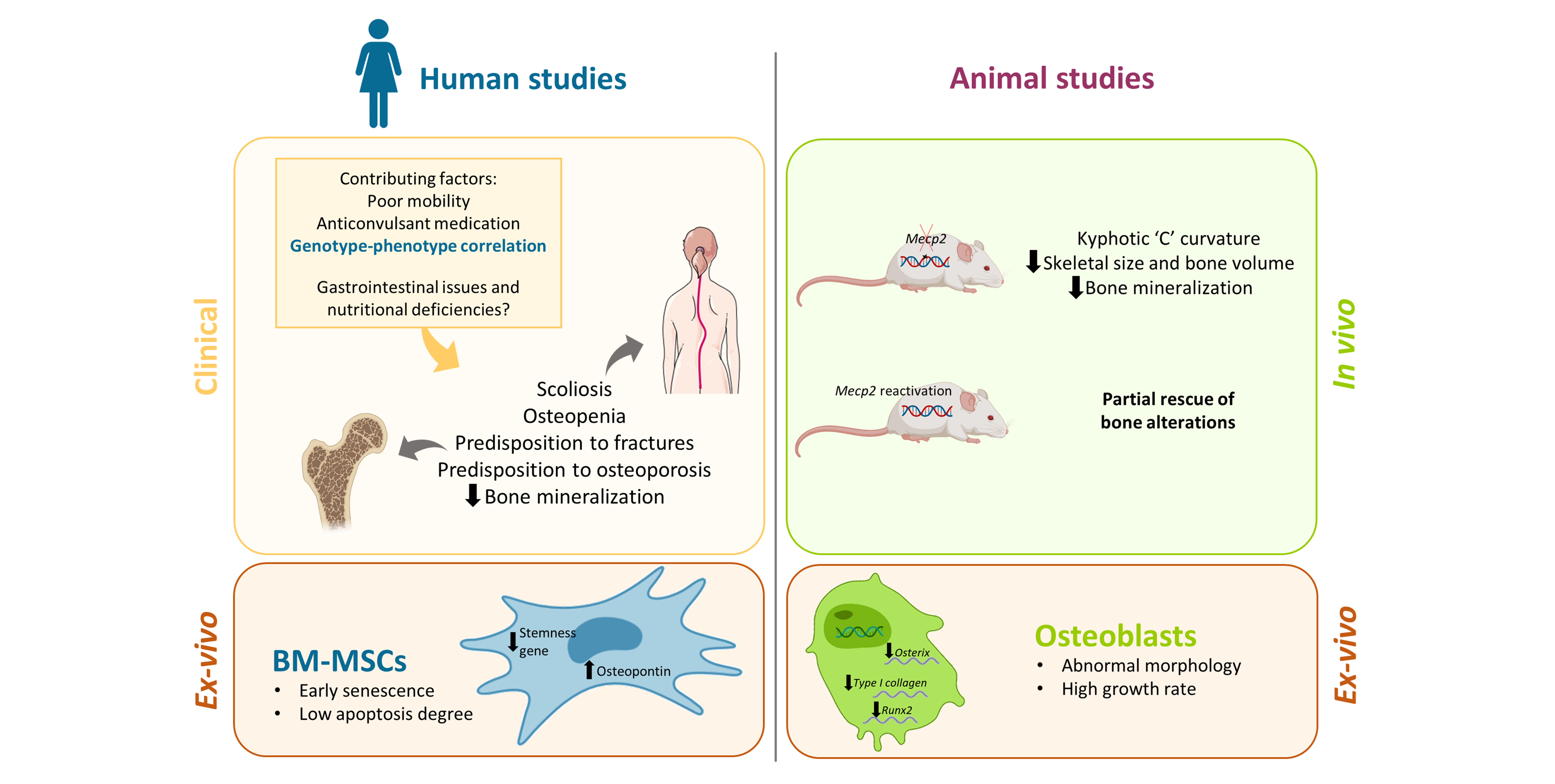
2. Clinical Aspects of Altered Bone and Mineral Metabolism in RTT
3. Mecp2 Deficiency Is Involved in the Impaired Bone Status in RTT: Evidence from Animal Models
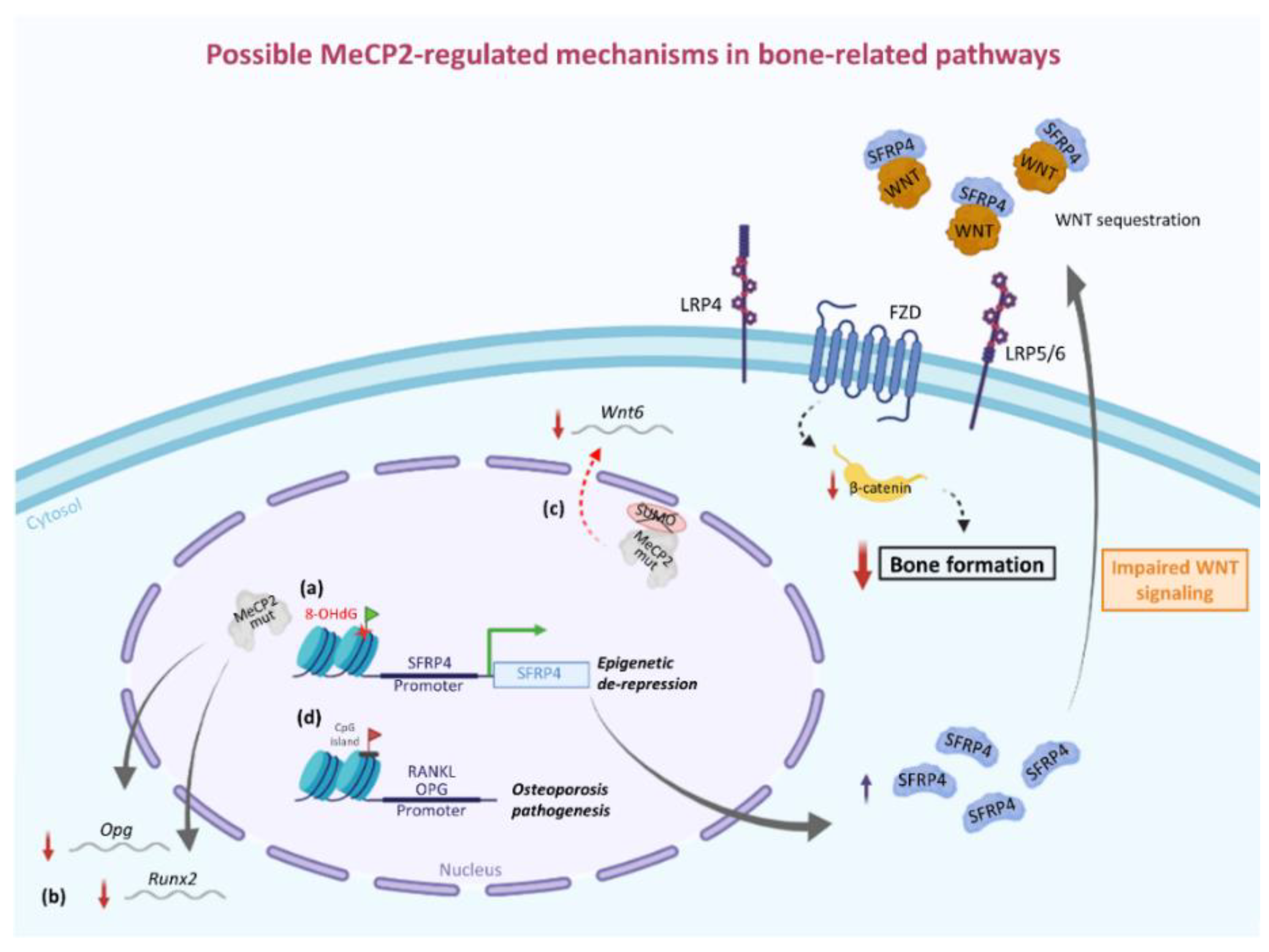
4. Involvement of MeCP2 in the Epigenetic Regulation of Bone-Related Factors: Possible Molecular/Cellular Mechanisms from Human and Animal Studies
5. Interactions of Bone with Other Organ Systems in RTT
6. Evidence from Other Neurodevelopmental Disabilities
7. Therapeutic Approaches for Bone-Related Issues in RTT
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonard, H.; Bower, C.; English, D. The Prevalence and Incidence of Rett Syndrome in Australia. Eur. Child Adolesc. Psychiatry 1997, 6, 8–10. [Google Scholar]
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett Syndrome in Australia: A Review of the Epidemiology. J. Pediatr. 2006, 148, 347–352. [Google Scholar] [CrossRef]
- Wong, V.C.N.; Li, S.Y.H. Rett Syndrome: Prevalence among Chinese and a Comparison of MECP2 Mutations of Classic Rett Syndrome with Other Neurodevelopmental Disorders. J. Child Neurol. 2007, 22, 1397–1400. [Google Scholar] [CrossRef]
- Fehr, S.; Bebbington, A.; Nassar, N.; Downs, J.; Ronen, G.M.; de Klerk, N.; Leonard, H. Trends in the Diagnosis of Rett Syndrome in Australia. Pediatr. Res. 2011, 70, 313–319. [Google Scholar] [CrossRef]
- Anderson, A.; Wong, K.; Jacoby, P.; Downs, J.; Leonard, H. Twenty Years of Surveillance in Rett Syndrome: What Does This Tell Us? Orphanet J. Rare Dis. 2014, 9, 87. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Rastegar, M. Rett Syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett Syndrome: MeCP2 Levels Display Tissue- and Cell-Specific Differences and Correlate with Neuronal Maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Signorini, C.; Leoncini, S.; De Felice, C.; Pecorelli, A.; Meloni, I.; Ariani, F.; Mari, F.; Amabile, S.; Paccagnini, E.; Gentile, M.; et al. Redox Imbalance and Morphological Changes in Skin Fibroblasts in Typical Rett Syndrome. Oxidative Med. Cell. Longev. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Song, C.; Feodorova, Y.; Guy, J.; Peichl, L.; Jost, K.L.; Kimura, H.; Cardoso, M.C.; Bird, A.; Leonhardt, H.; Joffe, B.; et al. DNA Methylation Reader MECP2: Cell Type- and Differentiation Stage-Specific Protein Distribution. Epigenetics Chromatin 2014, 7, 17. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N.; et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity 2015, 42, 679–691. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, C.M.; Lima, M.P.; Kaufmann, W.E.; Bressler, J.P. Methyl CpG Binding Protein 2 Deficiency Enhances Expression of Inflammatory Cytokines by Sustaining NF-ΚB Signaling in Myeloid Derived Cells. J. Neuroimmunol. 2015, 283, 23–29. [Google Scholar] [CrossRef]
- Li, Z.; Song, S.; Zha, S.; Wang, C.; Chen, S.; Wang, F. MeCP2 Promotes Endothelial-to-Mesenchymal Transition in Human Endothelial Cells by Downregulating BMP7 Expression. Exp. Cell Res. 2019, 375, 82–89. [Google Scholar] [CrossRef]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non–Cell Autonomous Influence of MeCP2-Deficient Glia on Neuronal Dendritic Morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.G.; Guyenet, P.G.; Kipnis, J. Wild-Type Microglia Arrest Pathology in a Mouse Model of Rett Syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MeCP2 Isoform-Specific Antibody Reveals the Endogenous MeCP2E1 Expression in Murine Brain, Primary Neurons and Astrocytes. PLoS ONE 2012, 7, e49763. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.B.; Rastegar, M. Brain Region-Specific Expression of MeCP2 Isoforms Correlates with DNA Methylation within Mecp2 Regulatory Elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 Update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J.; Grimm, A.; Maher, T.; Bennetts, B. RettBASE: The IRSA MECP2 Variation Database—a New Mutation Database in Evolution. Hum. Mutat. 2003, 21, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett Syndrome: A Neurological Disorder with Metabolic Components. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific Mutations in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-Binding Protein 2 (MECP2) Mutation Type Is Associated with Disease Severity in Rett Syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Makita, Y.; Ogawa, A.; Amamiya, S.; Yamamoto, M.; Miyamoto, A.; Oki, J. The Role of Different X-Inactivation Pattern on the Variable Clinical Phenotype with Rett Syndrome. Brain Dev. 2001, 23, S161–S164. [Google Scholar] [CrossRef][Green Version]
- Hoffbuhr, K.C.; Moses, L.M.; Jerdonek, M.A.; Naidu, S.; Hoffman, E.P. Associations BetweenmeCP2 Mutations, x-Chromosome Inactivation, and Phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 99–105. [Google Scholar] [CrossRef]
- Young, J.I.; Zoghbi, H.Y. X-Chromosome Inactivation Patterns Are Unbalanced and Affect the Phenotypic Outcome in a Mouse Model of Rett Syndrome. Am. J. Hum. Genet. 2004, 74, 511–520. [Google Scholar] [CrossRef]
- Knudsen, G.P.S.; Neilson, T.C.S.; Pedersen, J.; Kerr, A.; Schwartz, M.; Hulten, M.; Bailey, M.E.S.; Ørstavik, K.H. Increased Skewing of X Chromosome Inactivation in Rett Syndrome Patients and Their Mothers. Eur. J. Hum. Genet. 2006, 14, 1189–1194. [Google Scholar] [CrossRef]
- Glaze, D.G.; Frost, J.D.; Zoghbi, H.Y.; Percy, A.K. Rett’s Syndrome: Characterization of Respiratory Patterns and Sleep. Ann. Neurol. 1987, 21, 377–382. [Google Scholar] [CrossRef]
- Ellaway, C.; Sholler, G.; Leonard, H.; Christodoulou, J. Prolonged QT Interval in Rett Syndrome. Arch. Dis. Child 1999, 80, 470–472. [Google Scholar] [CrossRef][Green Version]
- Jefferson, A.; Leonard, H.; Siafarikas, A.; Woodhead, H.; Fyfe, S.; Ward, L.M.; Munns, C.; Motil, K.; Tarquinio, D.; Shapiro, J.R.; et al. Clinical Guidelines for Management of Bone Health in Rett Syndrome Based on Expert Consensus and Available Evidence. PLoS ONE 2016, 11, e0146824. [Google Scholar] [CrossRef]
- Tarquinio, D.C.; Hou, W.; Berg, A.; Kaufmann, W.E.; Lane, J.B.; Skinner, S.A.; Motil, K.J.; Neul, J.L.; Percy, A.K.; Glaze, D.G. Longitudinal Course of Epilepsy in Rett Syndrome and Related Disorders. Brain 2017, 140, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Tarquinio, D.C.; Hou, W.; Neul, J.L.; Berkmen, G.K.; Drummond, J.; Aronoff, E.; Harris, J.; Lane, J.B.; Kaufmann, W.E.; Motil, K.J.; et al. The Course of Awake Breathing Disturbances across the Lifespan in Rett Syndrome. Brain Dev. 2018, 40, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Faundez, V.; Wynne, M.; Crocker, A.; Tarquinio, D. Molecular Systems Biology of Neurodevelopmental Disorders, Rett Syndrome as an Archetype. Front. Integr. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Motil, K.J.; Barrish, J.O.; Neul, J.L.; Glaze, D.G. Low Bone Mineral Mass Is Associated with Decreased Bone Formation and Diet in Females with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 386–392. [Google Scholar] [CrossRef]
- Roende, G.; Petersen, J.; Ravn, K.; Fuglsang, K.; Andersen, H.; Nielsen, J.B.; Brøndum-Nielsen, K.; Jensen, J.-E.B. Low Bone Turnover Phenotype in Rett Syndrome: Results of Biochemical Bone Marker Analysis. Pediatr. Res. 2014, 75, 551–558. [Google Scholar] [CrossRef]
- Blue, M.E.; Boskey, A.L.; Doty, S.B.; Fedarko, N.S.; Hossain, M.A.; Shapiro, J.R. Osteoblast Function and Bone Histomorphometry in a Murine Model of Rett Syndrome. Bone 2015, 76, 23–30. [Google Scholar] [CrossRef]
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med. Wochenschr. 1966, 116, 723–726. [Google Scholar]
- Naidu, S.; Murphy, M.; Moser, H.W.; Rett, A. Rett Syndrome—Natural History in 70 Cases. Am. J. Med. Genet. 1986, 1, 61–72. [Google Scholar] [CrossRef]
- Keret, D.; Bassett, G.S.; Bunnell, W.P.; Marks, H.G. Scoliosis in Rett Syndrome. J. Pediatr. Orthop. 1988, 8, 138–142. [Google Scholar] [CrossRef]
- Bassett, G.S.; Tolo, V.T. The Incidence and Natural History of Scoliosis in Rett Syndrome. Dev. Med. Child Neurol. 1990, 32, 963–966. [Google Scholar] [CrossRef]
- Hennessy, M.J.; Haas, R.H. The Orthopedic Management of Rett Syndrome. J. Child Neurol. 1988, 3, S43–S47. [Google Scholar] [CrossRef]
- Holm, V.A.; King, H.A. Scoliosis in the Rett Syndrome. Brain Dev. 1990, 12, 151–153. [Google Scholar] [CrossRef]
- Huang, T.J.; Lubicky, J.P.; Hammerberg, K.W. Scoliosis in Rett Syndrome. Orthop. Rev. 1994, 23, 931–937. [Google Scholar]
- Loder, R.T.; Lee, C.L.; Richards, B.S. Orthopedic Aspects of Rett Syndrome: A Multicenter Review. J. Pediatr. Orthop. 1989, 9, 557–562. [Google Scholar] [CrossRef]
- Roberts, A.P.; Conner, A.N. Orthopaedic Aspects of Rett’s Syndrome: Brief Report. J. Bone Jt. Surg. Br. 1988, 70, 674. [Google Scholar] [CrossRef]
- Lidström, J.; Stokland, E.; Hagberg, B. Scoliosis in Rett Syndrome. Clinical and Biological Aspects. Spine 1994, 19, 1632–1635. [Google Scholar] [CrossRef]
- Killian, J.T.; Lane, J.B.; Lee, H.-S.; Skinner, S.A.; Kaufmann, W.E.; Glaze, D.G.; Neul, J.L.; Percy, A.K. Scoliosis in Rett Syndrome: Progression, Comorbidities, and Predictors. Pediatr. Neurol. 2017, 70, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.K.; Lee, H.-S.; Neul, J.L.; Lane, J.B.; Skinner, S.A.; Geerts, S.P.; Annese, F.; Graham, J.; McNair, L.; Motil, K.J.; et al. Profiling Scoliosis in Rett Syndrome. Pediatr. Res. 2010, 67, 435–439. [Google Scholar] [CrossRef]
- Haas, R.H.; Dixon, S.D.; Sartoris, D.J.; Hennessy, M.J. Osteopenia in Rett Syndrome. J. Pediatr. 1997, 131, 771–774. [Google Scholar] [CrossRef]
- Davies, J.H.; Evans, B.A.J.; Gregory, J.W. Bone Mass Acquisition in Healthy Children. Arch. Dis. Child 2005, 90, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Budden, S.S.; Gunness, M.E. Bone Histomorphometry in Three Females with Rett Syndrome. Brain Dev. 2001, 23, 133–137. [Google Scholar] [CrossRef]
- Budden, S.S.; Gunness, M.E. Possible Mechanisms of Osteopenia in Rett Syndrome: Bone Histomorphometric Studies. J. Child Neurol. 2003, 18, 698–702. [Google Scholar] [CrossRef]
- Leonard, H.; Thomson, M.; Bower, C.; Fyfe, S.; Constantinou, J. Skeletal Abnormalities in Rett Syndrome: Increasing Evidence for Dysmorphogenetic Defects. Am. J. Med. Genet. 1995, 58, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Thomson, M.; Glasson, E.; Fyfe, S.; Leonard, S.; Ellaway, C.; Christodoulou, J.; Bower, C. Metacarpophalangeal Pattern Profile and Bone Age in Rett Syndrome: Further Radiological Clues to the Diagnosis. Am. J. Med. Genet. 1999, 83, 88–95. [Google Scholar] [CrossRef]
- Leonard, H.; Thomson, M.R.; Glasson, E.J.; Fyfe, S.; Leonard, S.; Bower, C.; Christodoulou, J.; Ellaway, C. A Population-Based Approach to the Investigation of Osteopenia in Rett Syndrome. Dev. Med. Child Neurol. 1999, 41, 323–328. [Google Scholar] [CrossRef]
- Cepollaro, C.; Gonnelli, S.; Bruni, D.; Pacini, S.; Martini, S.; Franci, M.B.; Gennari, L.; Rossi, S.; Hayek, G.; Zappella, M.; et al. Dual X-Ray Absorptiometry and Bone Ultrasonography in Patients with Rett Syndrome. Calcif. Tissue Int. 2001, 69, 259–262. [Google Scholar] [CrossRef]
- Gonnelli, S.; Caffarelli, C.; Hayek, J.; Montagnani, A.; Cadirni, A.; Franci, B.; Lucani, B.; Rossi, S.; Nuti, R. Bone Ultrasonography at Phalanxes in Patients with Rett Syndrome: A 3-Year Longitudinal Study. Bone 2008, 42, 737–742. [Google Scholar] [CrossRef]
- Sarajlija, A.; Djuric, M.; Tepavcevic, D.K.; Grkovic, S.; Djordjevic, M. Vitamin D Deficiency in Serbian Patients with Rett Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E1972–E1978. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.R.; Bibat, G.; Hiremath, G.; Blue, M.E.; Hundalani, S.; Yablonski, T.; Kantipuly, A.; Rohde, C.; Johnston, M.; Naidu, S. Bone Mass in Rett Syndrome: Association with Clinical Parameters and MECP2 Mutations. Pediatr. Res. 2010, 68, 446–451. [Google Scholar] [CrossRef]
- Jefferson, A.; Fyfe, S.; Downs, J.; Woodhead, H.; Jacoby, P.; Leonard, H. Longitudinal Bone Mineral Content and Density in Rett Syndrome and Their Contributing Factors. Bone 2015, 74, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Caffarelli, C.; Francolini, V.; Hayek, J.; Valacchi, G.; Giannotti, S.; Nuti, R.; Gonnelli, S. Bone Status in Relation to Ambulatory Performance in Girls with Rett Syndrome: A 10-Year Longitudinal Study. Pediatr. Res. 2019, 85, 639–643. [Google Scholar] [CrossRef]
- Motil, K.J.; Schultz, R.J.; Abrams, S.; Ellis, K.J.; Glaze, D.G. Fractional Calcium Absorption Is Increased in Girls with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Motil, K.J.; Ellis, K.J.; Barrish, J.O.; Caeg, E.; Glaze, D.G. Bone Mineral Content and Bone Mineral Density Are Lower in Older than in Younger Females with Rett Syndrome. Pediatr. Res. 2008, 64, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Roende, G.; Ravn, K.; Fuglsang, K.; Andersen, H.; Nielsen, J.B.; Brøndum-Nielsen, K.; Jensen, J.-E.B. DXA Measurements in Rett Syndrome Reveal Small Bones with Low Bone Mass. J. Bone Miner. Res. 2011, 26, 2280–2286. [Google Scholar] [CrossRef]
- Caffarelli, C.; Gonnelli, S.; Pitinca, M.D.T.; Camarri, S.; Al Refaie, A.; Hayek, J.; Nuti, R. Methyl-CpG-Binding Protein 2 (MECP2) Mutation Type Is Associated with Bone Disease Severity in Rett Syndrome. BMC Med. Genet. 2020, 21. [Google Scholar] [CrossRef]
- Motil, K.J.; Caeg, E.; Barrish, J.O.; Geerts, S.; Lane, J.B.; Percy, A.K.; Annese, F.; McNair, L.; Skinner, S.A.; Lee, H.-S.; et al. Gastrointestinal and Nutritional Problems Occur Frequently Throughout Life in Girls and Women with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Motil, K.J.; Barrish, J.O.; Lane, J.; Geerts, S.P.; Annese, F.; McNair, L.; Percy, A.K.; Skinner, S.A.; Neul, J.L.; Glaze, D.G. Vitamin D Deficiency Is Prevalent in Females with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- O’Connor, R.D.; Zayzafoon, M.; Farach-Carson, M.C.; Schanen, N.C. Mecp2 Deficiency Decreases Bone Formation and Reduces Bone Volume in a Rodent Model of Rett Syndrome. Bone 2009, 45, 346–356. [Google Scholar] [CrossRef]
- Kamal, B.; Russell, D.; Payne, A.; Constante, D.; Tanner, K.E.; Isaksson, H.; Mathavan, N.; Cobb, S.R. Biomechanical Properties of Bone in a Mouse Model of Rett Syndrome. Bone 2015, 71, 106–114. [Google Scholar] [CrossRef]
- Ross, P.D.; Guy, J.; Selfridge, J.; Kamal, B.; Bahey, N.; Tanner, K.E.; Gillingwater, T.H.; Jones, R.A.; Loughrey, C.M.; McCarroll, C.S.; et al. Exclusive Expression of MeCP2 in the Nervous System Distinguishes between Brain and Peripheral Rett Syndrome-like Phenotypes. Hum. Mol. Genet. 2016, 25, 4389–4404. [Google Scholar] [CrossRef][Green Version]
- Squillaro, T.; Hayek, G.; Farina, E.; Cipollaro, M.; Renieri, A.; Galderisi, U. A Case Report: Bone Marrow Mesenchymal Stem Cells from a Rett Syndrome Patient Are Prone to Senescence and Show a Lower Degree of Apoptosis. J. Cell. Biochem. 2008, 103, 1877–1885. [Google Scholar] [CrossRef]
- Alvarez-Saavedra, M.; Carrasco, L.; Sura-Trueba, S.; Demarchi Aiello, V.; Walz, K.; Neto, J.X.; Young, J.I. Elevated Expression of MeCP2 in Cardiac and Skeletal Tissues Is Detrimental for Normal Development. Hum. Mol. Genet. 2010, 19, 2177–2190. [Google Scholar] [CrossRef] [PubMed]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.N.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT Signaling Promotes Osteogenesis by Directly Stimulating Runx2 Gene Expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Bonewald, L. The Role of the Wnt/β-Catenin Signaling Pathway in Formation and Maintenance of Bone and Teeth. Int. J. Biochem. Cell Biol. 2016, 77, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Huang, C.; Huang, Y.; Yang, Y.; He, X.; Zhang, L.; Lv, X.-W.; Jin, Y.; Li, J. MeCP2 Modulates the Canonical Wnt Pathway Activation by Targeting SFRP4 in Rheumatoid Arthritis Fibroblast-like Synoviocytes in Rats. Cell. Signal. 2013, 25, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Kitazawa, R.; Kondo, T.; Mori, M.; Hamada, Y.; Nishida, M.; Minami, Y.; Haraguchi, R.; Takahashi, Y.; Kitazawa, S. Diabetic Osteopenia by Decreased β-Catenin Signaling Is Partly Induced by Epigenetic Derepression of SFRP-4 Gene. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Kitazawa, S.; Haraguchi, R.; Kitazawa, R. Morphology-Oriented Epigenetic Research. Histochem. Cell Biol. 2018, 150, 3–12. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. SFRP4-Dependent Wnt Signal Modulation Is Critical for Bone Remodeling during Postnatal Development and Age-Related Bone Loss. Sci. Rep. 2016, 6, 25198. [Google Scholar] [CrossRef]
- Tai, D.J.C.; Liu, Y.C.; Hsu, W.L.; Ma, Y.L.; Cheng, S.J.; Liu, S.Y.; Lee, E.H.Y. MeCP2 SUMOylation Rescues Mecp2-Mutant-Induced Behavioural Deficits in a Mouse Model of Rett Syndrome. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Hsu, W.-L.; Ma, Y.-L.; Liu, Y.-C.; Tai, D.J.C.; Lee, E.H.Y. Restoring Wnt6 Signaling Ameliorates Behavioral Deficits in MeCP2 T158A Mouse Model of Rett Syndrome. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Kitazawa, R.; Kitazawa, S. Methylation Status of a Single CpG Locus 3 Bases Upstream of TATA-Box of Receptor Activator of Nuclear Factor-ΚB Ligand (RANKL) Gene Promoter Modulates Cell- and Tissue-Specific RANKL Expression and Osteoclastogenesis. Mol. Endocrinol. 2007, 21, 148–158. [Google Scholar] [CrossRef]
- Wang, P.; Cao, Y.; Zhan, D.; Wang, D.; Wang, B.; Liu, Y.; Li, G.; He, W.; Wang, H.; Xu, L. Influence of DNA Methylation on the Expression of OPG/RANKL in Primary Osteoporosis. Int. J. Med. Sci. 2018, 15, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Nectoux, J.; Fichou, Y.; Rosas-Vargas, H.; Cagnard, N.; Bahi-Buisson, N.; Nusbaum, P.; Letourneur, F.; Chelly, J.; Bienvenu, T. Cell Cloning-Based Transcriptome Analysis in Rett Patients: Relevance to the Pathogenesis of Rett Syndrome of New Human MeCP2 Target Genes. J. Cell. Mol. Med. 2010, 14, 1962–1974. [Google Scholar] [CrossRef]
- Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt, R.; Standridge, S.; Lane, J.; Dinkel, T.; Jones, M.; et al. Multisystem Comorbidities in Classic Rett Syndrome: A Scoping Review. BMJ Paediatr. Open 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Roth, C.; Christen, H.J.; Brockmann, K.; Hanefeld, F. Endocrinological Study on Growth Retardation in Rett Syndrome. Acta Paediatr. 2001, 90, 1257–1261. [Google Scholar] [CrossRef]
- Humphrey, K.N.; Horn, P.S.; Olshavsky, L.; Reebals, L.; Standridge, S.M. Features of Menstruation and Menstruation Management in Individuals with Rett Syndrome. J. Pediatr. Adolesc. Gynecol. 2021, 34, 144–153. [Google Scholar] [CrossRef]
- Kremer, R.; Gilsanz, V. Fat and Bone: An Odd Couple. Front. Endocrinol. 2016, 6. [Google Scholar] [CrossRef]
- Caffarelli, C.; Gonnelli, S.; Tanzilli, L.; Hayek, J.; Vichi, V.; Franci, M.B.; Lucani, B.; Nuti, R. The Relationship between Serum Ghrelin and Body Composition with Bone Mineral Density and QUS Parameters in Subjects with Rett Syndrome. Bone 2012, 50, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Delhanty, P.J.D.; van der Eerden, B.C.J.; van Leeuwen, J.P.T.M. Ghrelin and Bone. Biofactors 2014, 40, 41–48. [Google Scholar] [CrossRef]
- Kaji, H. Interaction between Muscle and Bone. J. Bone Metab. 2014, 21, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Liao, W. Psychomotor Dysfunction in Rett Syndrome: Insights into the Neurochemical and Circuit Roots. Dev. Neurobiol. 2019, 79, 51–59. [Google Scholar] [CrossRef]
- Conti, V.; Gandaglia, A.; Galli, F.; Tirone, M.; Bellini, E.; Campana, L.; Kilstrup-Nielsen, C.; Rovere-Querini, P.; Brunelli, S.; Landsberger, N. MeCP2 Affects Skeletal Muscle Growth and Morphology through Non Cell-Autonomous Mechanisms. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.L.; Christodoulou, J. Mitochondrial Dysfunction in the Skeletal Muscle of a Mouse Model of Rett Syndrome (RTT): Implications for the Disease Phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef]
- Sponseller, P.D.; Yazici, M.; Demetracopoulos, C.; Emans, J.B. Evidence Basis for Management of Spine and Chest Wall Deformities in Children. Spine 2007, 32, S81. [Google Scholar] [CrossRef]
- Cohen, J.L.; Klyce, W.; Kudchadkar, S.R.; Kotian, R.N.; Sponseller, P.D. Respiratory Complications after Posterior Spinal Fusion for Neuromuscular Scoliosis: Children With Rett Syndrome at Greater Risk Than Those With Cerebral Palsy. Spine 2019, 44, 1396–1402. [Google Scholar] [CrossRef]
- Rumbak, D.M.; Mowrey, W.; Schwartz, S.W.; Sarwahi, V.; Djukic, A.; Killinger, J.S.; Katyal, C. Spinal Fusion for Scoliosis in Rett Syndrome with an Emphasis on Respiratory Failure and Opioid Usage. J. Child Neurol. 2016, 31, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Karmaniolou, I.; Krishnan, R.; Galtrey, E.; Cleland, S.; Vijayaraghavan, R. Perioperative Management and Outcome of Patients with Rett Syndrome Undergoing Scoliosis Surgery: A Retrospective Review. J. Anesth. 2015, 29, 492–498. [Google Scholar] [CrossRef]
- Rettsyndrome.Org. Available online: https://www.rettsyndrome.org/ (accessed on 9 May 2021).
- Rett Syndrome Europe—RSE. Available online: https://www.rettsyndrome.eu/ (accessed on 9 May 2021).
- Downs, J.; Torode, I.; Wong, K.; Ellaway, C.; Elliott, E.J.; Izatt, M.T.; Askin, G.N.; Mcphee, B.I.; Cundy, P.; Leonard, H. Surgical Fusion of Early Onset Severe Scoliosis Increases Survival in Rett Syndrome: A Cohort Study. Dev. Med. Child Neurol. 2016, 58, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Whitney, D.G.; Caird, M.S.; Jepsen, K.J.; Kamdar, N.S.; Marsack-Topolewski, C.N.; Hurvitz, E.A.; Peterson, M.D. Elevated Fracture Risk for Adults with Neurodevelopmental Disabilities. Bone 2020, 130, 115080. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Hediger, M.L.; England, L.J.; Molloy, C.A.; Yu, K.F.; Manning-Courtney, P.; Mills, J.L. Reduced Bone Cortical Thickness in Boys with Autism or Autism Spectrum Disorder. J. Autism Dev. Disord. 2008, 38, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, A.M.; Cano Sokoloff, N.; McDonnell, E.; Macklin, E.A.; McDougle, C.J.; Misra, M. Bone Accrual in Males with Autism Spectrum Disorder. J. Pediatr. 2017, 181, 195–201. [Google Scholar] [CrossRef]
- Neumeyer, A.M.; O’Rourke, J.A.; Massa, A.; Lee, H.; Lawson, E.A.; McDougle, C.J.; Misra, M. Brief Report: Bone Fractures in Children and Adults with Autism Spectrum Disorders. J. Autism Dev. Disord. 2015, 45, 881–887. [Google Scholar] [CrossRef]
- Neumeyer, A.M.; Gates, A.; Ferrone, C.; Lee, H.; Misra, M. Bone Density in Peripubertal Boys with Autism Spectrum Disorders. J. Autism Dev. Disord. 2013, 43, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Roke, Y.; van Harten, P.N.; Buitelaar, J.K.; Tenback, D.E.; Quekel, L.G.B.A.; de Rijke, Y.B.; Boot, A.M. Bone Mineral Density in Male Adolescents with Autism Spectrum Disorders and Disruptive Behavior Disorder with or without Antipsychotic Treatment. Eur. J. Endocrinol. 2012, 167, 855–863. [Google Scholar] [CrossRef]
- Schreck, K.A.; Williams, K.; Smith, A.F. A Comparison of Eating Behaviors between Children with and without Autism. J. Autism Dev. Disord. 2004, 34, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Şengenç, E.; Kıykım, E.; Saltik, S. Vitamin D Levels in Children and Adolescents with Autism. J. Int. Med. Res. 2020, 48, 0300060520934638. [Google Scholar] [CrossRef] [PubMed]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal Symptoms in Autism Spectrum Disorder: A Meta-Analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef]
- Goodman, S.B.; Jiranek, W.; Petrow, E.; Yasko, A.W. The Effects of Medications on Bone. J. Am. Acad. Orthop. Surg. 2007, 15, 450–460. [Google Scholar] [CrossRef]
- Macdonald, M.; Esposito, P.; Ulrich, D. The Physical Activity Patterns of Children with Autism. BMC Res. Notes 2011, 4, 422. [Google Scholar] [CrossRef]
- Barnhill, K.; Ramirez, L.; Gutierrez, A.; Richardson, W.; Marti, C.N.; Potts, A.; Shearer, R.; Schutte, C.; Hewitson, L. Bone Mineral Density in Boys Diagnosed with Autism Spectrum Disorder: A Case-Control Study. J. Autism Dev. Disord. 2017, 47, 3608–3619. [Google Scholar] [CrossRef]
- Wren, T.A.L.; Kalkwarf, H.J.; Zemel, B.S.; Lappe, J.M.; Oberfield, S.; Shepherd, J.A.; Winer, K.K.; Gilsanz, V. Longitudinal Tracking of DXA Bone Measures Over 6 Years in Children and Adolescents: Persistence of Low Bone Mass to Maturity. J. Pediatr. 2014, 164, 1280–1285. [Google Scholar] [CrossRef]
- Ekhlaspour, L.; Baskaran, C.; Campoverde, K.J.; Sokoloff, N.C.; Neumeyer, A.M.; Misra, M. Bone Density in Adolescents and Young Adults with Autism Spectrum Disorders. J. Autism Dev. Disord. 2016, 46, 3387–3391. [Google Scholar] [CrossRef] [PubMed]
- Pitukcheewanont, P.; Chen, P. Bone Density Measurements in Children and Adolescents: Quantitative Computed Tomography versus Dual-Energy X-Ray Absorptiometry. Endocrinologist 2005, 15, 232–239. [Google Scholar] [CrossRef]
- Neumeyer, A.M.; Cano Sokoloff, N.; McDonnell, E.; Macklin, E.A.; McDougle, C.J.; Misra, M. Bone Microarchitecture in Adolescent Boys with Autism Spectrum Disorder. Bone 2017, 97, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; Howgate, D.; Anderson, W.; Howes, C.; Heliotis, M.; Mantalaris, A.; Tsiridis, E.; Tsapakis, E. Risk of Osteoporosis and Fracture Incidence in Patients on Antipsychotic Medication. Expert Opin. Drug Saf. 2011, 10, 575–602. [Google Scholar] [CrossRef]
- Motyl, K.J.; Dick-de-Paula, I.; Maloney, A.E.; Lotinun, S.; Bornstein, S.; de Paula, F.J.A.; Baron, R.; Houseknecht, K.L.; Rosen, C.J. Trabecular Bone Loss after Administration of the Second-Generation Antipsychotic Risperidone Is Independent of Weight Gain. Bone 2012, 50, 490–498. [Google Scholar] [CrossRef]
- Seriwatanachai, D.; Thongchote, K.; Charoenphandhu, N.; Pandaranandaka, J.; Tudpor, K.; Teerapornpuntakit, J.; Suthiphongchai, T.; Krishnamra, N. Prolactin Directly Enhances Bone Turnover by Raising Osteoblast-Expressed Receptor Activator of Nuclear Factor KappaB Ligand/Osteoprotegerin Ratio. Bone 2008, 42, 535–546. [Google Scholar] [CrossRef]
- Calarge, C.A.; Schlechte, J.A. Bone Health in Boys with Autism Spectrum Disorder. J. Autism Dev. Disord. 2017, 47, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.E.; Sharan, K.; Takumi, T.; Yadav, V.K. Skeletal Site-Specific Changes in Bone Mass in a Genetic Mouse Model for Human 15q11-13 Duplication Seen in Autism. Sci. Rep. 2017, 7, 9902. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, G.I.; Bertelloni, S. The Use of Bisphosphonates in Pediatrics. HRP 2014, 82, 290–302. [Google Scholar] [CrossRef]
- Caffarelli, C.; Hayek, J.; Nuti, R.; Gonnelli, S. Teriparatide in the Treatment of Recurrent Fractures in a Rett Patient. Clin. Cases Min. Bone Metab. 2015, 12, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Lotan, M.; Reves-Siesel, R.; Eliav-Shalev, R.S.; Merrick, J. Osteoporosis in Rett Syndrome: A Case Study Presenting a Novel Management Intervention for Severe Osteoporosis. Osteoporos Int. 2013, 24, 3059–3063. [Google Scholar] [CrossRef] [PubMed]
- Zanchetta, M.B.; Scioscia, M.F.; Zanchetta, J.R. Bone Microarchitecture in Rett Syndrome and Treatment with Teriparatide: A Case Report. Osteoporos Int. 2016, 27, 2873–2877. [Google Scholar] [CrossRef]
- Lambert, A.-S.; Rothenbuhler, A.; Charles, P.; Brailly-Tabard, S.; Trabado, S.; Célestin, E.; Durand, E.; Fontaine, I.; Miladi, L.; Wicart, P.; et al. Lower Incidence of Fracture after IV Bisphosphonates in Girls with Rett Syndrome and Severe Bone Fragility. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, A.; Renard, E.; Hernandez, M.; Dousset, B.; Brezin, F.; Lambert, L.; Weryha, G.; Feillet, F. Annual Injection of Zoledronic Acid Improves Bone Status in Children with Cerebral Palsy and Rett Syndrome. Calcif Tissue Int. 2019, 104, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.R.; Boskey, A.L.; Doty, S.B.; Lukashova, L.; Blue, M.E. Zoledronic Acid Improves Bone Histomorphometry in a Murine Model of Rett Syndrome. Bone 2017. [Google Scholar] [CrossRef]
- Shapiro, J.R.; Blue, M.E. Bisphosphonates and MeCP2 Deficiency: Cellular Studies and Clinical Application in Rett Syndrome. J. Musculoskelet Disord. Treat. 2018, 4. [Google Scholar] [CrossRef]
- Downs, J.; Bergman, A.; Carter, P.; Anderson, A.; Palmer, G.M.; Roye, D.; van Bosse, H.; Bebbington, A.; Larsson, E.L.; Smith, B.G.; et al. Guidelines for Management of Scoliosis in Rett Syndrome Patients Based on Expert Consensus and Clinical Evidence. Spine 2009, 34, E607–E617. [Google Scholar] [CrossRef]
- Lotan, M. Rett Syndrome. Guidelines for Individual Intervention. Sci. World J. 2006, 6, 1504–1516. [Google Scholar] [CrossRef]
- Fonzo, M.; Sirico, F.; Corrado, B. Evidence-Based Physical Therapy for Individuals with Rett Syndrome: A Systematic Review. Brain Sci. 2020, 10, 410. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.Y.; Wender, A.R.; Jones, M.D.; Fung, E.B.; Pico, E.L. The Effect of Low Magnitude Mechanical Stimulation (LMMS) on Bone Density in Patients with Rett Syndrome: A Pilot and Feasibility Study. J. Pediatr. Rehabil. Med. 2014, 7, 167–178. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Alteration of Bone Status | Compartment/ Bone Investigated | Genotype- Phenotype Correlation | Other Contributing Factors | References | |
|---|---|---|---|---|---|
| Scoliosis | Spine | Yes | - | [37,38,39,40,41,42,43,44,45,46,47,48,65] | |
| Fracture predisposition ↑ osteoporosis risk | ↓ bone mineralization | Whole body | - | - | [49] |
| Femur and total hip | Yes (MECP2 mutations R106T, R168X, R255X R270X) | - | [65] | ||
| Osteopenia | ↓ bone volume ↓ bone formation rate | Iliac crest | - | - | [51,52] |
| ↓ bone formation rate ↔ bone resorption | Whole body | - | Diet | [34,35] | |
| ↓ cortical thickness ↓ % cortical area | Metacarpal bone | - | Age Anticonvulsant medication | [55] | |
| ↓ bone mineral density (BMD) ↓ bone quality | Radius, heel by Achilles, phalanges | - | Anticonvulsant medication Vitamin D levels Ambulatory status | [56,57] | |
| ↓ bone mineral content and density | Whole body | No | Ambulatory status Age Seizures Anticonvulsant medication Mild hypercalciuria | [60,62,63,64] | |
| ↓ bone area and lean tissue mass | Whole body | - | Ambulatory status | [60] | |
| Worse bone properties | Phalanges | - | Ambulatory status | [61] | |
| ↓ bone mineral density (BMD) | Femur and total hip Lumbar spine | Yes (MECP2 mutations R106T, R168X, R255X R270X, T158M) | - | [59,65] | |
| Dysmorphogenetic defect | Metatarsal and metacarpal shortness Short distal phalanx of the thumb | Hands and feet | - | - | [53,54] |
| Animal Models | Bone-Related Hallmarks | Compartment/Bone/Cells Investigated for Bone Status | References |
|---|---|---|---|
| Mecp2-/y mice | Kyphotic ‘C’ curvature | Spine | [36,59,69] |
| Short femurs | Femur | ||
| ↓ skeletal size | |||
| ↓ cortical and trabecular bone | Femur | ||
| ↓ mineral apposition rate | Femur and calvarial bone | ||
| ↔ osteoblast and osteoclast counts | Femur and tibia | ||
| ↔ levels of OPG and RANKL | Serum | ||
| ↓ cortical thickness and medullary mineralization | Long bones and spine | ||
| Mecp2stop/y male and Mecp2+/stop female mice | ↓ weight and length | Femur and tibia | [70] |
| ↓ cortical bone stiffness, micro-hardness, and tensile modulus | Tibia | ||
| ↓ collagen content | Femur | ||
| Altered trabecular bone architecture | Femur and vertebrae (L5) | ||
| ↔ osteoclast counts | Femur | ||
| Mecp2-null males and heterozygous females | ↓ mineral apposition rate ↓ mineralizing surface ↓ bone formation rate/bone surface ↓ osteoblast number Abnormal morphology of osteoblasts ↑ growth rate ↓ Osterix, Runx2, and type I collagen mRNA levels | Femoral trabecular and calvarial bone Femur Femur Femur and tibia Femur and tibia Femur-derived osteoblasts Femur-derived osteoblasts Femur-derived osteoblasts | [36] |
| Mecp2 peripheral knockout mice | ↓ strength, hardness, and fracture threshold | Tibia | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecorelli, A.; Cordone, V.; Schiavone, M.L.; Caffarelli, C.; Cervellati, C.; Cerbone, G.; Gonnelli, S.; Hayek, J.; Valacchi, G. Altered Bone Status in Rett Syndrome. Life 2021, 11, 521. https://doi.org/10.3390/life11060521
Pecorelli A, Cordone V, Schiavone ML, Caffarelli C, Cervellati C, Cerbone G, Gonnelli S, Hayek J, Valacchi G. Altered Bone Status in Rett Syndrome. Life. 2021; 11(6):521. https://doi.org/10.3390/life11060521
Chicago/Turabian StylePecorelli, Alessandra, Valeria Cordone, Maria Lucia Schiavone, Carla Caffarelli, Carlo Cervellati, Gaetana Cerbone, Stefano Gonnelli, Joussef Hayek, and Giuseppe Valacchi. 2021. "Altered Bone Status in Rett Syndrome" Life 11, no. 6: 521. https://doi.org/10.3390/life11060521
APA StylePecorelli, A., Cordone, V., Schiavone, M. L., Caffarelli, C., Cervellati, C., Cerbone, G., Gonnelli, S., Hayek, J., & Valacchi, G. (2021). Altered Bone Status in Rett Syndrome. Life, 11(6), 521. https://doi.org/10.3390/life11060521