
Emerging Treatment Options for Multi-Drug-Resistant Bacterial Infections

 ,
,  ,
, 

Abstract
1. Introduction
2. Clinically Relevant Multi-Drug-Resistant Bacteria and Their Main Resistance Mechanisms/Determinants
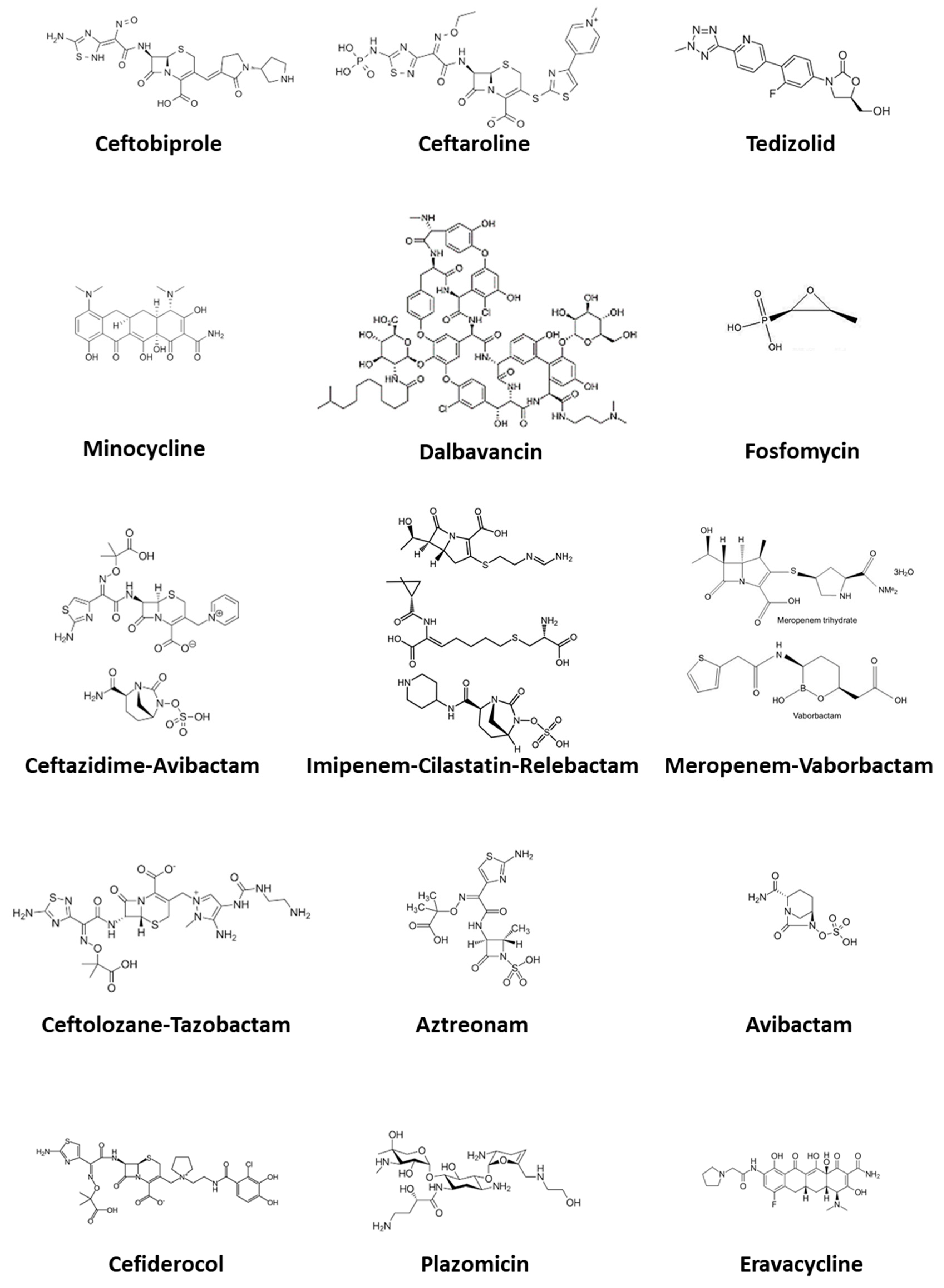
3. Emerging Antimicrobial Options for MDR Gram-Positive Cocci
3.1. Ceftobiprole
3.2. Ceftaroline
3.3. Dalbavancin
3.4. Tedizolid
3.5. Fosfomycin Disodium
4. Emerging Antimicrobial Options for MDR Gram-Negative Rods
4.1. Ceftolozane-Tazobactam
4.2. Ceftazidime-Avibactam
4.3. Meropenem-Vaborbactam
4.4. Imipenem-Cilastatin-Relebactam
4.5. Plazomicin
4.6. Minocycline
4.7. Aztreonam
4.8. Aztreonam-Avibactam
4.9. Cefiderocol
4.10. Eravacycline
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Statements
References
- Durante Mangoni, E. Parsimonious use of antibiotics in COVID-19: A missed opportunity. Clin. Infect. Immun. 2021, 6, 29–30. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Bush, K.; Harbarth, S.; Paul, M.; Rex, J.H.; Tacconelli, E.; Thwaites, G.E. Critical analysis of antibacterial agents in clinical development. Nat. Rev. Microbiol. 2020, 18, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Xu, E.; Song, A.; Sum Mok, I.Y.; Karageorgopoulos, D.E.; Armaganidis, A.; Tsiodras, S.; Lipman, J. Emerging treatment options for infections by multidrug-resistant gram-positive microorganisms. Microorganisms 2020, 8, 191. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Abbott, I.J.; Peleg, A.Y. Stenotrophomonas, achromobacter, and nonmelioid burkholderia species: Antimicrobial resistance and therapeutic strategies. Semin. Respir. Crit. Care Med. 2015, 36, 99–110. [Google Scholar] [CrossRef]
- Pompilio, A.; Piccolomini, R.; Picciani, C.; D’Antonio, D.; Savini, V.; Di Bonaventura, G. Factors associated with adherence to and biofilm formation on polystyrene by stenotrophomonas maltophilia: Role of cell surface hydrophobicity and mortality. FEMS Microbiol. Lett. 2008. [Google Scholar] [CrossRef]
- Denton, M.; Kerr, K.G. Microbiological and clinical aspects of infection associated with stenotrophomonas maltophilia. Clin. Microbiol. Rev. 1998. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Urban, T.; Goldberg, J. The multifarious, multireplicon Burkholderia cepaciaepacian complex. Nat. Rev. Microbiol. 2005. [Google Scholar] [CrossRef]
- Isler, B.; Kidd, T.J.; Stewart, A.G.; Harris, P.; Paterson, D.L. Achromobacter infections and treatment options. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No eskape! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.S. Which antibiotic for resistant gram-positives, and why? J. Infect. 2014, 68. [Google Scholar] [CrossRef]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Schneider, T.; Sahl, H.-G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 139–158. [Google Scholar] [CrossRef]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of antibiotic resistance in important gram-positive and gram-negative pathogens and novel antibiotic solutions. Antibiotics 2021, 10, 415. [Google Scholar] [CrossRef]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in Enterococci. Exp. Rev. Anti Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Miller, W.R.; Murray, B.E.; Rice, L.B.; Arias, C.A. Resistance in vancomycin-resistant Enterococci. Infect. Dis. Clin. N. Am. 2020, 34, 751–771. [Google Scholar] [CrossRef]
- Cherazard, R.; Epstein, M.; Doan, T.L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial resistant Streptococcus pneumoniae: Prevalence, mechanisms, and clinical implications. Am. J. Ther. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Eichenberger, E.M.; Thaden, J.T. Epidemiology and mechanisms of resistance of extensively drug resistant gram-negative bacteria. Antibiotics 2019, 8, 37. [Google Scholar] [CrossRef]
- Bush, B.; Jacoby, G.; Medeiros, M. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1997, 39, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Philippon, A.; Jacquier, H.; Ruppé, H.; Labia, R. Structure-based classification of class A beta-lactamases. Curr. Res. Transl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Poirel, L. Epidemiology and diagnostics of carbapenem resistance in gram-negative bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef] [PubMed]
- Giacobbe, D.R.; De Rosa, F.G.; Del Bono, V.; Grossi, P.A.; Pea, F.; Petrosillo, N. Ceftobiprole: Drug evaluation and place in therapy. Exp. Rev. Anti Infect. Ther. 2019, 17, 689–698. [Google Scholar] [CrossRef]
- Pfaller, M.; Flamm, R.K.; Duncan, L.R.; Shortridge, D.; Smart, J.I.; Hamed, K.A.; Mendes, R.; Sader, H.S. Ceftobiprole activity when tested against contemporary bacteria causing bloodstream infections in the United States (2016–2017). Diagn. Microbiol. Infect. Dis. 2019, 94, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Bloem, A.; Bax, H.; Erlangga, Y.; Verkaik, N. New-generation antibiotics for treatment of gram-positive infections: A review with focus on endocarditis and osteomyelitis. J. Clin. Med. 2021, 10, 1743. [Google Scholar] [CrossRef] [PubMed]
- Backstrom, T.; Panagiotidis, G.; Beck, O.; Asker-Hagelberg, C.; Rashid, M.; Weintraub, A.; Nord, C. Effect of ceftobiprole on the normal human intestinal microflora. Int. J. Antimicrob. Agents 2010, 36, 537–541. [Google Scholar] [CrossRef]
- Scheeren, T.W.L. Ceftobiprole medocaril in the treatment of hospital-acquired pneumonia. Future Microbiol. 2015, 10, 1913–1928. [Google Scholar] [CrossRef]
- Campanile, F.; Bongiorno, D.; Mongelli, G.; Zanghì, G.; Stefani, S. Bactericidal activity of ceftobiprole combined with different antibiotics against selected gram positive isolates. Diagn. Microbiol. Infect. Dis. 2019, 93, 77–81. [Google Scholar] [CrossRef] [PubMed]
- White, B.P.; Barber, K.E.; Stover, K.R. Ceftaroline for the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Am. J. Health Syst. Pharm. 2017, 74, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, C.; Crum-Cianflone, N.F. Ceftaroline: A new cephalosporin with activity against methicillin-resistant Staphylococcus aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3. [Google Scholar] [CrossRef]
- Justo, J.A.; Mayer, S.M.; Pai, M.P.; Soriano, M.M.; Danziger, L.H.; Novak, R.M.; Rodvold, K.A. Pharmacokinetics of ceftaroline in normal body weight and obese (classes I, II, and III) healthy adult subjects. Antimicrob. Agents Chemother. 2015, 59, 3956–3965. [Google Scholar] [CrossRef]
- European Medicines Agency. Zinforo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zinforo (accessed on 1 June 2021).
- Corey, G.R.; Wilcox, M.; Talbot, G.H.; Friedland, H.D.; Baculik, T.; Witherell, G.W.; Critchley, I.; Das, A.F.; Thye, D. Integrated analysis of CANVAS 1 and 2: Phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin. Infect. Dis. 2010, 51, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Assisa, L.; Nedeljković, M.; Dessena, A. New strategies for targeting and treatment of multi-drug resistant Staphylococcus aureus. Drug Resist. Updat. 2017, 31, 1–14. [Google Scholar] [CrossRef]
- Smith, J.R.; Roberts, K.D.; Rybak, M.J. Dalbavancin: A novel lipoglycopeptide antibiotic with extended activity against gram-positive infections. Infect. Dis. Ther. 2015, 4, 245–258. [Google Scholar] [CrossRef]
- Bouza, E.; Valerio, M.; Soriano, A.; Morata, L.; Carus, E.G.; Rodríguez-González, M.C.; Hidalgo-Tenorio, C.; Plata, A.; Muñoz, P.; Vena, A.; et al. Dalbavancin in the treatment of different gram-positive infections: A real-life experience. Int. J. Antimicrob. Agents 2018, 51, 571–577. [Google Scholar] [CrossRef]
- Wang, J.; Wang, R.; Li, Y.; Cai, Y. Efficacy and safety of dalbavancin in the treatment of gram-positive bacterial infections. J. Glob. Antimicrob. Resist. 2021, 24, 72–80. [Google Scholar] [CrossRef]
- Flanagan, S.; Fang, E.; Muñoz, K.; Minassian, S.; Prokocimer, F. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy. 2014, 34, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Ferrández, O.; Urbina, O.; Grau, S. Critical role of tedizolid in the treatment of acute bacterial skin and skin structure infections. Drug Des. Dev. Ther. 2016, 11, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Carvalhaes, C.G.; Sader, H.S.; Flamm, R.K.; Streit, J.M.; Mendes, R.E. Assessment of tedizolid in vitro activity and resistance mechanisms against a collection of enterococcus spp. causing invasive infections, including isolates requiring an optimized dosing strategy for daptomycin from U.S. and European medical centers, 2016 to 2018. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Kullar, R.; Puzniak, L.A.; Swindle, J.P.; Lodise, T. Retrospective real-world evaluation of outcomes in patients with skin and soft structure infections treated with tedizolid in an outpatient setting. Infect. Dis. Ther. 2020, 9, 107–117. [Google Scholar] [CrossRef]
- Moran, G.J.; Fang, E.; Corey, G.R.; Das, A.F.; De Anda, C.; Prokocimer, P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): A randomized, double-blind, phase 3, non-inferiority trial. Lancet Infect. Dis. 2014, 14, 696–705. [Google Scholar] [CrossRef]
- Falagas, M.E.; Vouloumanou, E.K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Legakis, N.J.; Triarides, N.; Falagas, M.E. Susceptibility of contemporary isolates to fosfomycin: A systematic review of the literature. Int. J. Antimicrob. Agents 2016, 47, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.; Doi, Y. Fosfomycin: Resurgence of an old companion. J. Infect. Chemother. 2016, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Candel, F.J.; Cantón, R. Current approach to fosfomycin: From bench to bedside. Enferm. Infecc. Microbiol. Clin. 2019, 37, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Hsu, A.J. Defining the role of novel β-lactam agents that target carbapenem-resistant gram-negative organisms. J. Pediatr. Infect. Dis. Soc. 2019, 8, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, D.; Bonomo, R.A. Ceftazidime/avibactam and ceftolozane/tazobactam: “Second generation” beta-Lactam/beta lactamase combinations. Clin. Infect. Dis. 2016, 63, 234–241. [Google Scholar] [CrossRef]
- Wright, H.; Bonomo, R.; Paterson, D. New agents for the treatment of infections with gram-negative bacteria: Restoring the miracle or false dawn? Clin. Microbiol. Infect. 2017, 23, 704–712. [Google Scholar] [CrossRef]
- Skalweit, M. Profile of ceftolozane/tazobactam and its potential in the treatment of complicated intra-abdominal infections. Drug Des. Dev. Ther. 2015, 9, 2919–2925. [Google Scholar] [CrossRef]
- Wagenlehner, F.M.; Umeh, O.; Steenbergen, J.; Yuan, G.; Darouiche, R.O. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: A randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet 2015, 385, 1949–1956. [Google Scholar] [CrossRef]
- Huntington, J.A.; Sakoulas, G.; Umeh, O.; Cloutier, D.J.; Steenbergen, J.N.; Bliss, C.; Goldstein, E.J.C. Efficacy of ceftolozane/tazobactam versus levofloxacin in the treatment of complicated urinary tract infections (cUTIs) caused by levofloxacinresistant pathogens: Results from the ASPECT-cUTI trial. J. Antimicrob. Chemother. 2016, 71, 2014–2021. [Google Scholar] [CrossRef]
- Solomkin, J.; Hershberger, E.; Miller, B.; Popejoy, M.; Friedland, I.; Steenbergen, J.; Yoon, M.; Collins, S.; Yuan, G.; Barie, P.S.; et al. Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: Results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin. Infect. Dis. 2015, 60, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Zinforo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zavicefta (accessed on 1 June 2021).
- Castòn, J.J.; Lacort-Peralta, I.; Martín-Dávila, P.; Loeches, B.; Tabares, S.; Temkin, L.; Torre-Cisneros, J.; Paño-Pardo, J.R. Clinical efficacy of ceftazidime/avibactam versus other active agents for the treatment of bacteremia due to carbapenemase-producing Enterobacteriaceae in hematologic patients. Int. J. Infect. Dis. 2017, 59, 118–123. [Google Scholar] [CrossRef]
- Shields, R.K.; Potoski, B.A.; Haidar, G.; Hao, B.; Doi, Y.; Chen, L.; Press, E.G.; Kreiswirth, B.N.; Clancy, C.J. Clinical outcomes, drug toxicity, and emergence of ceftazidime-avibactam resistance among patients treated for carbapenem-resistant Enterobacteriaceae infections. J. Antimicrob. Chemother. 2016, 71, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Tuon, F.F.; Rocha, J.L.; Formigoni-Pinto, M.R. Pharmacological aspects and spectrum of action of ceftazidime–avibactam: A systematic review. Infection 2018, 46, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Temkin, E.; Torre-Cisneros, J.; Beovic, B.; Benito, N.; Giannella, M.; Gilarranz, R.; Jeremiah, C.; Loeches, B.; Machuca, I.; Jiménez-Martín, M.J.; et al. Ceftazidime-avibactam as salvage therapy for infections caused by carbapenem-resistant organisms. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Zasowski, E.J.; Rybak, J.M.; Rybak, M.J. The beta-lactams strike back: Ceftazidime-avibactam. Pharmacotherapy 2015, 35, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Hemarajata, P.; Sun, D.; Rubio-Aparicio, D.; Tsivkovski, R.; Yang, S.; Sebra, R.; Kasarskis, A.; Nguyen, H.; Hanson, B.M.; et al. Resistance to ceftazidime-avibactam is due to transposition of KPC in a porin-deficient strain of Klebsiella pneumoniae with increased efflux activity. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Van Duin, D.; Lok, J.J.; Earley, M.; Cober, E.; Richter, S.S.; Perez, F.; Salata, R.A.; Kalayjian, R.C.; Watkins, R.R.; Doi, Y.; et al. Colistin versus ceftazidime-avibactam in the treatment of infections due to carbapenem-resistant Enterobacteriaceae. Clin. Infect. Dis. 2018, 66, 163–171. [Google Scholar] [CrossRef]
- Rodriguez-Bano, J.; Gutiérrez-Gutiérrez, B.; Machuca, I.; Pascual, A. Treatment of infections caused by extended-spectrum-beta-lactamase-, ampc-, and carbapenemase-producing Enterobacteriaceae. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Pogue, J.M.; Bonomo, R.A.; Kaye, K.S. Ceftazidime/avibactam, meropenem/vaborbactam, or both? Clinical and formulary considerations. Clin. Infect. Dis. 2019, 68, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Huband, M.D.; Mendes, R.E.; Flamm, R.K.; Castanheira, M. In vitro activity of meropenem/vaborbactam and characterisation of carbapenem resistance mechanisms among carbapenem-resistant Enterobacteriaceae from the 2015 meropenem/vaborbactam surveillance programme. Int. J. Antimicrob. Agents 2018, 52, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kaye, K.S.; Bhowmick, T.; Metallidis, S.; Bleasdale, S.C.; Sagan, O.S.; Stus, V.; Vazquez, J.; Zaitsev, V.; Bidair, M.; Chorvat, E.; et al. Effect of meropenem-vaborbactam vs. piperacillin-tazobactam on clinical cure or improvement and microbial eradication in complicated urinary tract infection: The TANGO I randomized clinical trial. JAMA 2018, 319, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Wunderink, R.G.; Giamarellos-Bourboulis, E.J.; Rahav, G.; Mathers, A.J.; Bassetti, M.; Vazquez, J.; Cornely, O.A.; Solomkin, J.; Bhowmick, T.; Bishara, J.; et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: The TANGO II randomized clinical trial. Infect. Dis. Ther. 2018, 7, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Rybak, J.M.; Claeys, K.C. Imipenem-cilastatin-relebactam: A novel β-Lactam–β-Lactamase inhibitor combination for the treatment of multidrug-resistant gram-negative infections. Pharmacotherapy 2020, 40, 343–356. [Google Scholar] [CrossRef]
- Lob, S.H.; Hackel, M.A.; Kazmierczak, K.M.; Hoban, D.J.; Young, K.; Motyl, M.R.; Karlowsky, J.A.; Sahm, D.F. In vitro activity of imipenem-relebactam against gram-negative bacilli isolated from patients with lower respiratory tract infections in the United States in 2015—Results from the SMART global surveillance program. Diagn. Microbiol. Infect. Dis. 2017, 88, 171–176. [Google Scholar] [CrossRef]
- Lucasti, C.; Vasile, L.; Sandesc, D.; Venskutonis, D.; McLeroth, P.; Lala, M.; Rizk, M.L.; Brown, M.L.; Losada, M.C.; Pedley, A.; et al. Phase 2, dose-ranging study of relebactam with imipenem-cilastatin in subjects with complicated intra-abdominal infection. Antimicrob. Agents Chemother. 2016, 60, 6234–6243. [Google Scholar] [CrossRef]
- Wu, J.; Racine, F.; Wismer, M.K.; Young, K.; Carr, D.M.; Xiao, J.C.; Katwaru, R.; Si, Q.; Harradine, P.; Motyl, M.; et al. Exploring the pharmacokinetic/pharmacodynamic relationship of relebactam (MK-7655) in combination with imipenem in a hollow-fiber infection model. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Bhagunde, P.; Zhang, Z.; Racine, F.; Carr, D.; Wu, J.; Young, K.; Rizk, M.L. A translational pharmacokinetic/pharmacodynamic model to characterize bacterial kill in the presence of imipenem-relebactam. Int. J. Infect. Dis. 2019, 89, 55–61. [Google Scholar] [CrossRef]
- Motsch, J.; De Oliveira, C.M.; Stus, V.; Köksal, I.; Lyulko, O.; Boucher, H.W.; Kaye, K.S.; File, T.M.; Brown, M.L.; Khan, I.; et al. RESTORE-IMI 1: A multicenter, randomized, double-blind trial comparing efficacy and safety of imipenem/relebactam vs. colistin plus imipenem in patients with imipenem-nonsusceptible bacterial infections. Clin. Infect. Dis. 2020, 70, 1799–1808. [Google Scholar] [CrossRef]
- Titov, I.; Wunderink, R.G.; Roquilly, A.; Gonzalez, D.R.; David-Wang, A.; Boucher, H.W.; Kaye, K.S.; Losada, M.C.; Du, J.; Tipping, R.; et al. A randomized, double-blind, multicenter trial comparing efficacy and safety of imipenem/cilastatin/relebactam versus piperacillin/tazobactam in adults with hospital-acquired or ventilator-associated bacterial pneumonia (RESTORE-IMI 2 study). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Almaghrabi, R.; Clancy, C.J.; Doi, Y.; Hao, B.; Chen, L.; Shields, R.K.; Press, E.G.; Iovine, N.M.; Townsend, B.M.; Wagener, M.M.; et al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob. Agents Chemother. 2014, 58, 4443–4451. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Paul, M. Developing a new antibiotic for extensively drug-resistant pathogens: The case of plazomicin. Clin. Microbiol. Infect. 2018, 24, 1231–1233. [Google Scholar] [CrossRef]
- Wagenlehner, F.M.E.; Cloutier, D.J.; Komirenko, A.S.; Cebrik, D.S.; Krause, K.M.; Keepers, T.R.; Connolly, L.E.; Miller, L.G.; Friedland, I.; Dwyer, J.P. Once-daily plazomicin for complicated urinary tract infections. N. Engl. J. Med. 2019, 380, 729–740. [Google Scholar] [CrossRef]
- McKinnell, J.A.; Dwyer, J.P.; Talbot, G.H.; Connolly, L.E.; Friedland, I.; Smith, A.; Jubb, A.M.; Serio, A.W.; Krause, K.M.; Daikos, G.L. Plazomicin for infections caused by carbapenem-resistant Enterobacteriaceae. N. Engl. J. Med. 2019, 380, 791–793. [Google Scholar] [CrossRef]
- Shaeer, K.M.; Zmarlicka, M.T.; Chahine, E.B.; Piccicacco, N.; Cho, J.C. Plazomicin: A next-generation aminoglycoside. Pharmacotherapy 2019, 39, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; El Chakhtoura, N.G.; Papp-Wallace, K.M.; Wilson, B.M.; Bonomo, R.A. Treatment options for infections caused by carbapenem-resistant Enterobacteriaceae: Can we apply “precision medicine” to antimicrobial chemotherapy? Exp. Opin. Pharmacother. 2016, 17, 761–781. [Google Scholar] [CrossRef]
- Pogue, J.M.; Neelakanta, A.; Mynatt, R.P.; Sharma, S.; Lephart, P.; Kaye, K.S. Carbapenem-resistance in gram-negative bacilli and intravenous minocycline: An antimicrobial stewardship approach at the Detroit Medical Center. Clin. Infect. Dis. 2014, 59. [Google Scholar] [CrossRef]
- Zhou, J.; Ledesma, K.R.; Chang, K.T.; Abodakpi, H.; Gao, S.; Tam, V.H. Pharmacokinetics and pharmacodynamics of minocycline against Acinetobacter baumannii in a neutropenic murine pneumonia model. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Smith, K.P.; Whitfield, B.A.; Zucchi, P.C.; Lasco, T.M.; Bias, T.E.; Kirby, J.E.; Hirsch, E.B. Activity of minocycline against Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae clinical isolates, with comparison to doxycycline and tigecycline. Diagn. Microbiol. Infect. Dis. 2017, 88, 365–367. [Google Scholar] [CrossRef]
- Durante-Mangoni, E.; Utili, R.; Zarrilli, R. Combination therapy in severe Acinetobacter baumannii infections: An update on the evidence to date. Future Microbiol. 2014, 9, 773–789. [Google Scholar] [CrossRef]
- Colton, B.; McConeghy, K.W.; Schreckenberger, P.C.; Danziger, L.H. IV minocycline revisited for infections caused by multidrug-resistant organisms. Am. J. Health Syst. Pharm. 2016, 73, 279–285. [Google Scholar] [CrossRef]
- Greig, S.L.; Scott, L.J. Intravenous minocycline: A review in acinetobacter infections. Drugs 2016, 76, 1467–1476. [Google Scholar] [CrossRef]
- Cozzani, E.; Agnoletti, A.; Riva, S.; Parodi, A. Minocycline: A new molecule inducing subacute cutaneous lupus erythematosus? G. Ital. Dermatol. Venereol. 2015, 150, 261–265. [Google Scholar]
- Emeraud, C.; Escaut, L.; Boucly, A.; Fortineau, N.; Bonnin, R.A.; Naas, T.; Dortet, L. Aztreonam plus clavulanate, tazobactam, or avibactam for treatment of infections caused by metallo-beta-lactamase-producing gram-negative bacteria. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.B. Inhaled antibiotics for ventilator-associated infections. Infect. Dis. Clin. N. Am. 2017, 31, 577–591. [Google Scholar] [CrossRef]
- Ramsey, C.; MacGowan, A.P. A review of the pharmacokinetics and pharmacodynamics of aztreonam. J. Antimicrob. Chemother. 2016, 71, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, I.; Galani, I.; Papoutsaki, V.; Galani, L.; Giamarellou, H. Carbapenemase producing Klebsiella pneumoniae: Implication on future therapeutic strategies. Exp. Rev. Anti Infect. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Karlowsky, J.A.; Kazmierczak, K.M.; de Jonge, B.L.M.; Hackel, M.A.; Sahm, D.F.; Bradford, P.A. In vitro activity of aztreonam-avibactam against Enterobacteriaceae and Pseudomonas aeruginosa isolated by clinical laboratories in 40 countries from 2012 to 2015. Antimicrob. Agent Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Carvalhaes, C.G.; Arends, S.J.R.; Castanheira, M.; Mendes, R.E. Aztreonam/avibactam activity against clinical isolates of Enterobacterales collected in Europe, Asia and Latin America in 2019. J. Antimicrob. Chemother. 2021, 76, 659–666. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A.; et al. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant gram-negative bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef]
- Bassetti, M.; Echols, R.; Matsunaga, Y.; Ariyasu, M.; Doi, Y.; Ferrer, R.; Lodise, T.P.; Naas, T.; Niki, Y.; Paterson, D.L.; et al. Efficacy and safety of cefiderocol or best available therapy for the treatment of serious infections caused by carbapenem-resistant Gram-negative bacteria (CREDIBLE-CR): A randomised, open-label, multicentre, pathogen-focused, descriptive, phase 3 trial. Lancet Infect. Dis. 2021, 21, 226–240. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Baxter, M.R.; Adam, H.J.; Sutcliffe, J.; Karlowsky, J.A. In vitro activity of eravacycline against 2213 gram-negative and 2424 gram-positive bacterial pathogens isolated in Canadian hospital laboratories: CANWARD surveillance study 2014–2015. Diagn. Microbiol. Infect. Dis. 2018, 91, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Burton, C.E. Eravacycline, a newly approved fluorocycline. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1787–1794. [Google Scholar] [CrossRef]
- Yusuf, E.; Bax, H.; Verkaik, N.; van Westreenen, M. An update on eight “new” antibiotics against multidrug-resistant gram-negative bacteria. J. Clin. Med. 2021, 10, 1068. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Eravacycline: A review in complicated intra-abdominal infections. Drugs 2019, 79, 315–324. [Google Scholar] [CrossRef]
- WHO. WHO—Global Shortage of Innovative Antibiotics Fuels Emergence and Spread of Drug-Resistance. Departmental News, 15 April 2021. Available online: https://www.who.int/news/item/15-04-2021-global-shortage-of-innovative-antibiotics-fuels-emergence-and-spread-of-drug-resistance (accessed on 1 June 2021).


{kind=link}
| Mechanism(s) of Resistance | Current Options | Emerging Options | |
|---|---|---|---|
| GRAM-POSITIVE COCCI | |||
| Methicillin-resistant staphylococci (MRSA, MRCoNS) | PBP2a expression | Daptomycin, Linezolid | Ceftaroline, Ceftobiprole, Tedizolid, Dalbavancin, Fosfomycin |
| Vancomycin intermediate Staphylococcus aureus (VISA) | Chromosomal mutations | Daptomycin, Linezolid | Tedizolid, Dalbavancin |
| Vancomycin resistant Staphylococcus aureus (VRSA) | vanA gene expression | Daptomycin, Linezolid | Tedizolid, Dalbavancin |
| Ampicillin-resistant enterococci (ARE) | PBP mutation/overexpression | Vancomycin, Linezolid, Daptomycin | Dalbavancin, Ceftobiprole |
| Vancomycin-resistant enterococci (VRE) | vanA, vanB gene expression | Linezolid, Daptomycin | Tedizolid, Dalbavancin |
| Penicillin-resistant Streptococcus pneumoniae (PRSP) | PBP mutation | Ceftriaxone | Ceftaroline, Ceftobiprole, Tedizolid |
| GRAM-NEGATIVE BACILLI | |||
| Carbapenem-resistant Enterobacterales | β-lactamase production | Colistin, Tigecycline | KPC/OXA-48: Ceftazidime-Avibactam KPC: Meropenem-Vaborbactam KPC: Imipenem-Relebactam MBL: Aztreonam-Avibactam Fosfomycin Eravacycline Plazomicin Cefiderocol |
| XDR Pseudomonas aeruginosa | β-lactamase production Porin loss/mutation Efflux pump expression | Colistin | Ceftolozane-Tazobactam Ceftazidime-Avibactam Aztreonam/Aztreonam-Avibactam Fosfomycin Cefiderocol |
| Acinetobacter baumannii | β-lactamase production Porin loss/mutation Efflux pump expression | Colistin, Tigecycline | Cefiderocol Minocycline Eravacycline |
| Stenotrophomonas malthophilia/Burkolderia cepacia | β-lactamase production Porin loss/mutation Efflux pump expression | Co-trimoxazole | Cefiderocol Ceftazidime-Avibactam |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giurazza, R.; Mazza, M.C.; Andini, R.; Sansone, P.; Pace, M.C.; Durante-Mangoni, E. Emerging Treatment Options for Multi-Drug-Resistant Bacterial Infections. Life 2021, 11, 519. https://doi.org/10.3390/life11060519
Giurazza R, Mazza MC, Andini R, Sansone P, Pace MC, Durante-Mangoni E. Emerging Treatment Options for Multi-Drug-Resistant Bacterial Infections. Life. 2021; 11(6):519. https://doi.org/10.3390/life11060519
Chicago/Turabian StyleGiurazza, Roberto, Maria Civita Mazza, Roberto Andini, Pasquale Sansone, Maria Caterina Pace, and Emanuele Durante-Mangoni. 2021. "Emerging Treatment Options for Multi-Drug-Resistant Bacterial Infections" Life 11, no. 6: 519. https://doi.org/10.3390/life11060519
APA StyleGiurazza, R., Mazza, M. C., Andini, R., Sansone, P., Pace, M. C., & Durante-Mangoni, E. (2021). Emerging Treatment Options for Multi-Drug-Resistant Bacterial Infections. Life, 11(6), 519. https://doi.org/10.3390/life11060519