
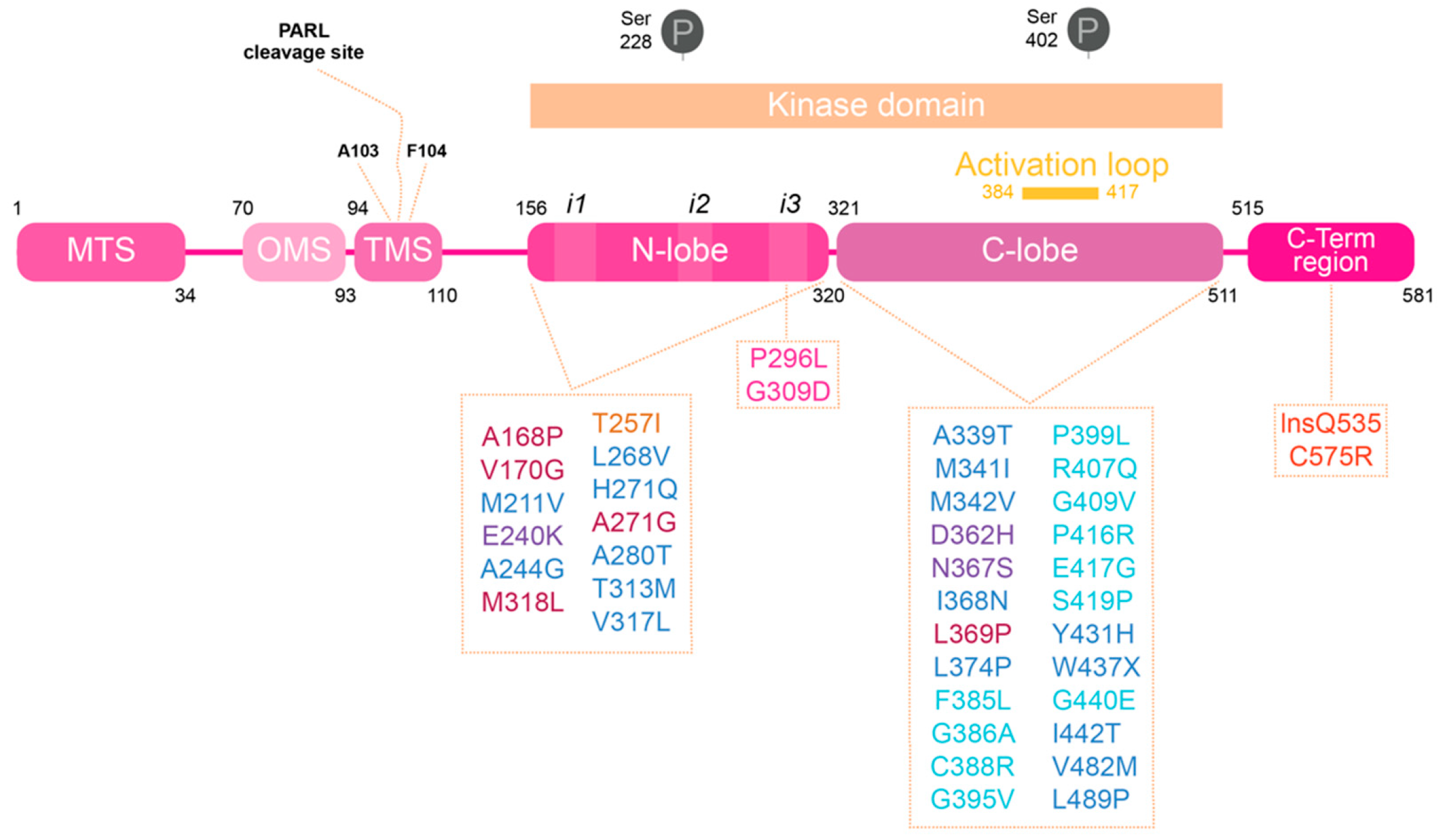
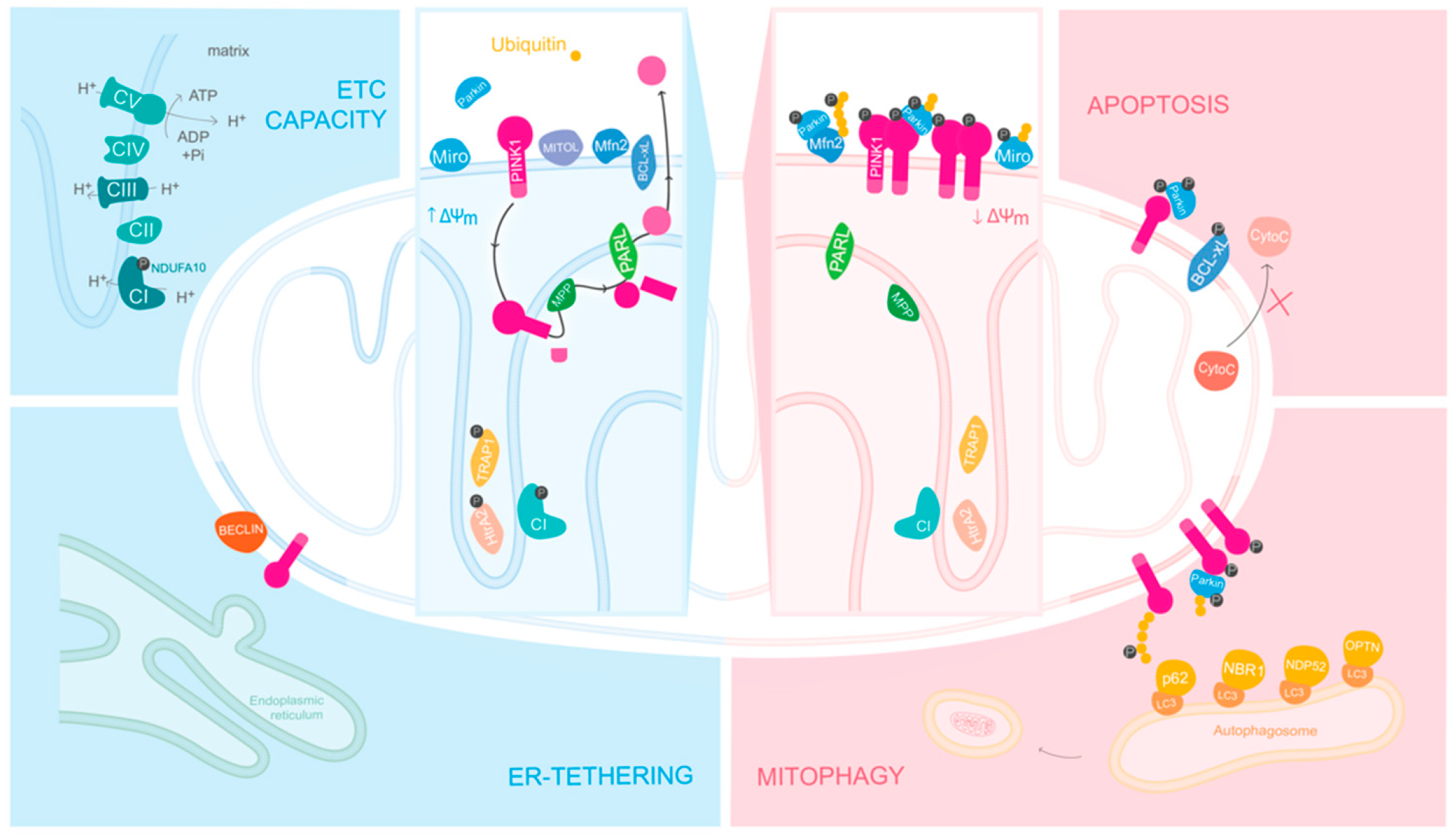
PINK1: A Bridge between Mitochondria and Parkinson’s Disease


{kind=link}
{kind=link}
Abstract
Share and Cite
Gonçalves, F.B.; Morais, V.A. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life 2021, 11, 371. https://doi.org/10.3390/life11050371
Gonçalves FB, Morais VA. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life. 2021; 11(5):371. https://doi.org/10.3390/life11050371
Chicago/Turabian StyleGonçalves, Filipa Barroso, and Vanessa Alexandra Morais. 2021. "PINK1: A Bridge between Mitochondria and Parkinson’s Disease" Life 11, no. 5: 371. https://doi.org/10.3390/life11050371
APA StyleGonçalves, F. B., & Morais, V. A. (2021). PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life, 11(5), 371. https://doi.org/10.3390/life11050371