Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Kinases on the Outer Mitochondrial Membrane
3. Phosphorylation in Mitochondrial Import Machinery
4. Kinases and Metabolism
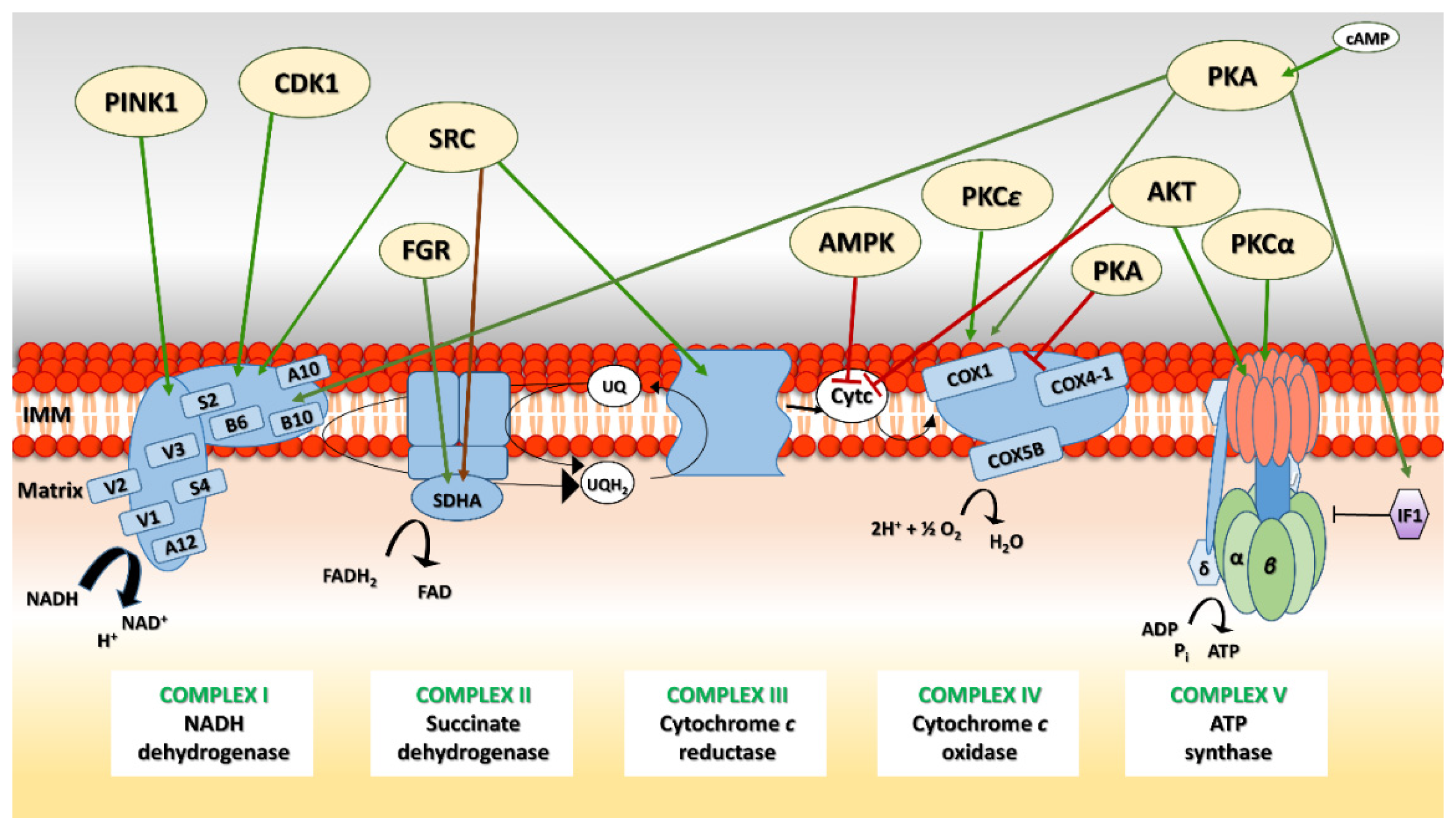
5. OXPHOS System Modification
6. Mitochondrial Quality Control
7. Nucleoid and Ribosomes (mtDNA Maintenance, Transcription and Protein Synthesis)
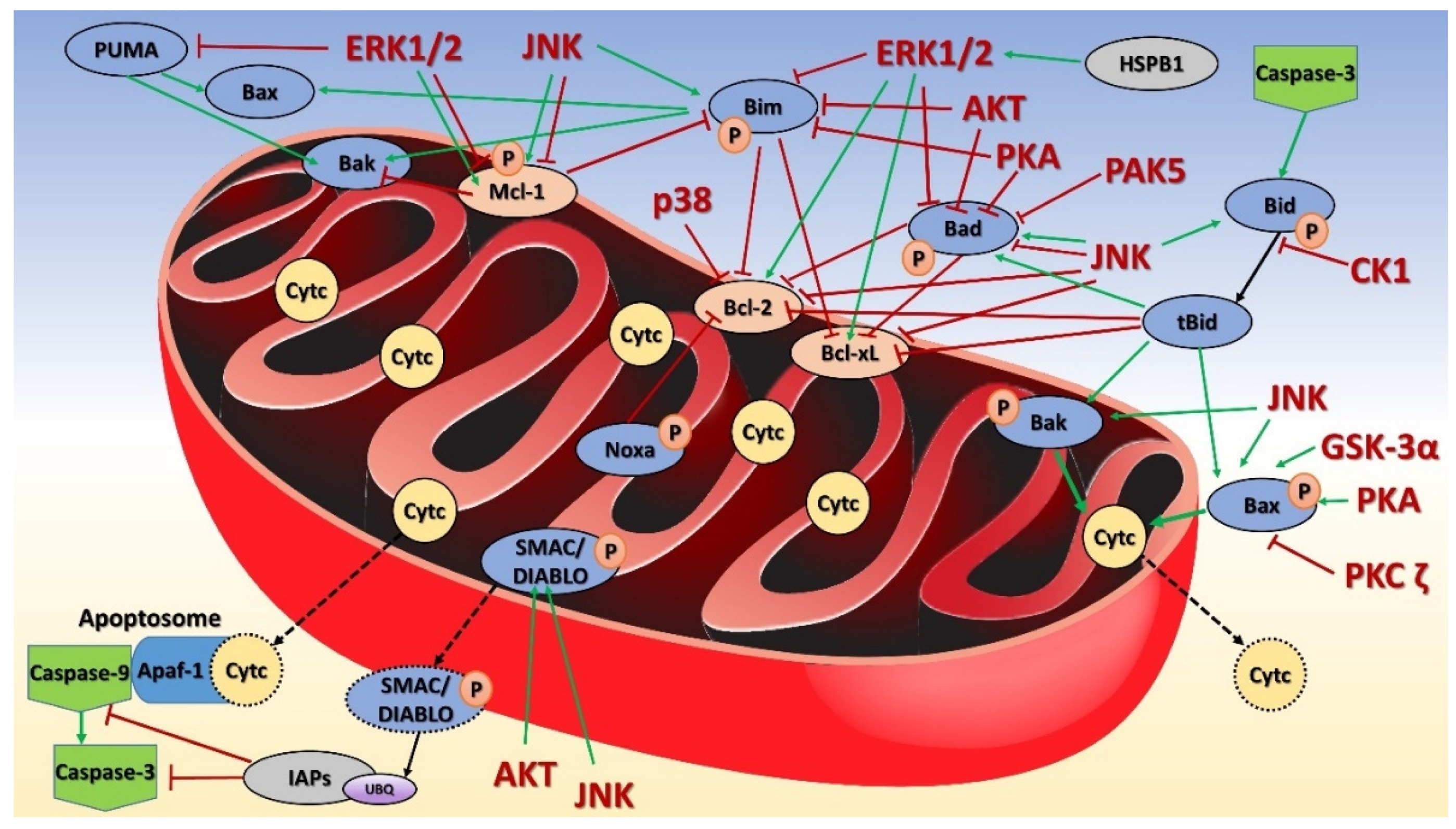
8. Mitochondrial Kinases in Apoptosis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AAC | ADP/ATP carrier protein |
| Abf2 | ARS-binding factor 2 |
| ABL | Abelson tyrosine kinase |
| ACC2 | acetyl-CoA carboxylase 2 |
| AD | Alzheimer’s disease |
| AKAP | A-kinase anchoring protein |
| AKT/PKB | protein kinase B |
| AMPK | 5′-AMP-activated protein kinase |
| BAT | brown adipose tissue |
| CAMK | Ca2+/calmodulin-dependent protein kinase |
| CDK | cyclin-dependent protein kinase |
| CK | casein kinase |
| COX | cytochrome c oxidase |
| CREB | cAMP-response element binding protein |
| Cytc | cytochrome c |
| DBP | dodecamer-binding protein |
| DRP1 | dynamin-related protein 1 |
| EF-Tu | translational elongation factor |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular receptor kinase 1/2 |
| ETC | electron transport chain |
| FDH | formate dehydrogenase |
| GSK-3β | glycogen synthase kinase 3β |
| HSP | heat-shock protein |
| IF | inhibitory factor |
| IMM | inner mitochondrial membrane |
| HIF | hypoxia-inducible transcription factor |
| HMG | high-mobility group |
| JNK | Jun N-terminal kinase |
| LRRK2 | leucine-rich repeat kinase 2 |
| MAPK | mitogen-activated protein kinase |
| MFF | mitochondrial fission factor |
| Mfn | mitofusin |
| MFP1 | MAR-binding filament-like protein 1 |
| MIA | mitochondrial intermembrane space assembly |
| MIEF1/2 | mitochondrial elongation factor 1/2 |
| MIM | mitochondrial import complex |
| MOMP | mitochondrial outer membrane permeabilization |
| MPP | mitochondrial processing peptidase |
| MPQC | mitochondrial protein quality control |
| mTERF1 | mitochondrial termination factor 1 |
| MTF1 | mitochondrial transcription factor 1 |
| mTOR | mammalian target of rapamycin |
| mtSSB | mitochondrial single-stranded DNA-binding protein |
| mtUPR | mitochondrial unfolded protein response |
| OMM | outer mitochondrial membrane |
| OXPHOS | oxidative phosphorylation |
| PAK5 | p21-activated kinase 5 |
| PARL | presenilins-associated rhomboid-like protein |
| PD | Parkinson’s disease |
| PDC | pyruvate dehydrogenase complex |
| PDH | pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| PDP | pyruvate dehydrogenase phosphatase |
| PGK1 | phosphoglycerate kinase 1 |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKR | protein kinase R |
| PINK1 | PTEN-induced putative kinase 1 |
| PLK3 | Polo-like kinase 3 |
| POLRMT | mitochondrial RNA polymerase |
| PTM | post-translational modification |
| ROS | reactive-oxygen species |
| SAM | sorting and assembly machinery |
| SAPK | stress-induced protein kinase |
| SDH | succinate dehydrogenase |
| SDHA | succinate dehydrogenase complex subunit A |
| sHSP | small heat-shock protein |
| StAR | steroidogenic acute regulatory protein |
| TCA | tricarboxylic acid cycle |
| TFAM | mitochondrial transcription factor A |
| TFB2M | mitochondrial transcription factor B |
| TFD | trophic factor deprivation |
| TIM | translocase of the inner mitochondrial membrane |
| TNFα | tumor necrosis factor-α |
| TOM | translocase of the outer mitochondrial membrane |
| TPKI | Tau protein kinase I |
| TRAP1 | TNF receptor-associated protein 1 |
| UCP1 | uncoupling protein 1 |
| VSM | vascular smooth muscle |
| XIAP | X-linked inhibitor-of-apoptosis protein. |
References
- Kruse, R.; Hojlund, K. Mitochondrial phosphoproteomics of mammalian tissues. Mitochondrion 2017, 33, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Giorgianni, F.; Koirala, D.; Weber, K.T.; Beranova-Giorgianni, S. Proteome analysis of subsarcolemmal cardiomyocyte mitochondria: A comparison of different analytical platforms. Int. J. Mol. Sci. 2014, 15, 9285–9301. [Google Scholar] [CrossRef] [PubMed]
- Padrao, A.I.; Vitorino, R.; Duarte, J.A.; Ferreira, R.; Amado, F. Unraveling the phosphoproteome dynamics in mammal mitochondria from a network perspective. J. Proteome Res. 2013, 12, 4257–4267. [Google Scholar] [CrossRef] [PubMed]
- Lucero, M.; Suarez, A.E.; Chambers, J.W. Phosphoregulation on mitochondria: Integration of cell and organelle responses. CNS Neurosci. Ther. 2019, 25, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Dixon, J.E. Mitochondrial modulation: Reversible phosphorylation takes center stage? Trends Biochem. Sci. 2006, 31, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Brunati, A.M.; Toninello, A. Tyrosine phosphorylation in mitochondria: A new frontier in mitochondrial signaling. Free Radic. Biol. Med. 2005, 38, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Gambardella, J.; Fiordelisi, A.; Trimarco, B.; Ciccarelli, M.; Iaccarino, G.; Santulli, G. Mechanistic Role of Kinases in the Regulation of Mitochondrial Fitness. Adv. Exp. Med. Biol. 2017, 982, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Corum, D.G.; Tsichlis, P.N.; Muise-Helmericks, R.C. AKT3 controls mitochondrial biogenesis and autophagy via regulation of the major nuclear export protein CRM-1. FASEB J. 2014, 28, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Shaerzadeh, F.; Motamedi, F.; Khodagholi, F. Inhibition of akt phosphorylation diminishes mitochondrial biogenesis regulators, tricarboxylic acid cycle activity and exacerbates recognition memory deficit in rat model of Alzheimer’s disease. Cell Mol. Neurobiol. 2014, 34, 1223–1233. [Google Scholar] [CrossRef]
- Gerbeth, C.; Mikropoulou, D.; Meisinger, C. From inventory to functional mechanisms: Regulation of the mitochondrial protein import machinery by phosphorylation. FEBS J. 2013, 280, 4933–4942. [Google Scholar] [CrossRef]
- Opalinska, M.; Meisinger, C. Mitochondrial protein import under kinase surveillance. Microb. Cell 2014, 1, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lorenzo, P.S.; Bogi, K.; Blumberg, P.M.; Yuspa, S.H. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol. Cell Biol. 1999, 19, 8547–8558. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Bakajsova, D.; Clifton, G.L. Protein kinase C-epsilon modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am. J. Physiol. Renal Physiol. 2004, 286, F307–316. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kitagishi, Y.; Kobayashi, M. Function and characteristics of PINK1 in mitochondria. Oxid Med. Cell Longev. 2013, 2013, 601587. [Google Scholar] [CrossRef] [PubMed]
- Plun-Favreau, H.; Hardy, J. PINK1 in mitochondrial function. Proc. Natl. Acad. Sci. USA 2008, 105, 11041–11042. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakano, N.; Ogino, M.; Ichimura, M.; Minami, A.; Matsuda, S. PINK1 signaling in mitochondrial homeostasis and in aging (Review). Int. J. Mol. Med. 2017, 39, 3–8. [Google Scholar] [CrossRef]
- Debattisti, V.; Gerencser, A.A.; Saotome, M.; Das, S.; Hajnoczky, G. ROS Control Mitochondrial Motility through p38 and the Motor Adaptor Miro/Trak. Cell Rep. 2017, 21, 1667–1680. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Weindel, C.G.; Bell, S.L.; Vail, K.J.; West, K.O.; Patrick, K.L.; Watson, R.O. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. eLife 2020, 9. [Google Scholar] [CrossRef]
- Kumar, S.; Bharti, A.; Mishra, N.C.; Raina, D.; Kharbanda, S.; Saxena, S.; Kufe, D. Targeting of the c-Abl tyrosine kinase to mitochondria in the necrotic cell death response to oxidative stress. J. Biol. Chem. 2001, 276, 17281–17285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017, 8, e3117. [Google Scholar] [CrossRef] [PubMed]
- Koc, E.C.; Miller-Lee, J.L.; Koc, H. Fyn kinase regulates translation in mammalian mitochondria. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 533–540. [Google Scholar] [CrossRef]
- Djeungoue-Petga, M.A.; Lurette, O.; Jean, S.; Hamel-Cote, G.; Martin-Jimenez, R.; Bou, M.; Cannich, A.; Roy, P.; Hebert-Chatelain, E. Intramitochondrial Src kinase links mitochondrial dysfunctions and aggressiveness of breast cancer cells. Cell Death Dis. 2019, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E.; Jose, C.; Gutierrez Cortes, N.; Dupuy, J.W.; Rocher, C.; Dachary-Prigent, J.; Letellier, T. Preservation of NADH ubiquinone-oxidoreductase activity by Src kinase-mediated phosphorylation of NDUFB10. Biochim. Biophys. Acta 2012, 1817, 718–725. [Google Scholar] [CrossRef]
- Ogura, M.; Yamaki, J.; Homma, M.K.; Homma, Y. Mitochondrial c-Src regulates cell survival through phosphorylation of respiratory chain components. Biochem. J. 2012, 447, 281–289. [Google Scholar] [CrossRef]
- Gringeri, E.; Carraro, A.; Tibaldi, E.; D’Amico, F.E.; Mancon, M.; Toninello, A.; Pagano, M.A.; Vio, C.; Cillo, U.; Brunati, A.M. Lyn-mediated mitochondrial tyrosine phosphorylation is required to preserve mitochondrial integrity in early liver regeneration. Biochem. J. 2009, 425, 401–412. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Carrascoso, I.; Baixauli, F.; Roche-Molina, M.; Latorre-Pellicer, A.; Fernandez-Silva, P.; Mittelbrunn, M.; Sanchez-Madrid, F.; Perez-Martos, A.; Lowell, C.A.; et al. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab. 2014, 19, 1020–1033. [Google Scholar] [CrossRef]
- Salvi, M.; Morrice, N.A.; Brunati, A.M.; Toninello, A. Identification of the flavoprotein of succinate dehydrogenase and aconitase as in vitro mitochondrial substrates of Fgr tyrosine kinase. FEBS Lett. 2007, 581, 5579–5585. [Google Scholar] [CrossRef]
- Tibaldi, E.; Brunati, A.M.; Massimino, M.L.; Stringaro, A.; Colone, M.; Agostinelli, E.; Arancia, G.; Toninello, A. Src-Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J. Cell Biochem. 2008, 104, 840–849. [Google Scholar] [CrossRef]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Smith, K.R.; Lim, S.T.; Tian, R.; Lu, J.; Tan, M. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 2016, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Y.; Moss, S.B.; Rubin, C.S. Characterization of S-AKAP84, a novel developmentally regulated A kinase anchor protein of male germ cells. J. Biol. Chem. 1995, 270, 27804–27811. [Google Scholar] [CrossRef]
- Wiltshire, C.; Matsushita, M.; Tsukada, S.; Gillespie, D.A.; May, G.H. A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem. J. 2002, 367, 577–585. [Google Scholar] [CrossRef]
- Chambers, J.W.; Pachori, A.; Howard, S.; Iqbal, S.; LoGrasso, P.V. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J. Biol. Chem. 2013, 288, 4000–4011. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Bonestroo, H.J.; Zijlstra, J.; Kavelaars, A.; Heijnen, C.J. Mitochondrial JNK phosphorylation as a novel therapeutic target to inhibit neuroinflammation and apoptosis after neonatal ischemic brain damage. Neurobiol. Dis. 2013, 54, 432–444. [Google Scholar] [CrossRef]
- Court, N.W.; Kuo, I.; Quigley, O.; Bogoyevitch, M.A. Phosphorylation of the mitochondrial protein Sab by stress-activated protein kinase 3. Biochem. Biophys. Res. Commun. 2004, 319, 130–137. [Google Scholar] [CrossRef]
- Affaitati, A.; Cardone, L.; de Cristofaro, T.; Carlucci, A.; Ginsberg, M.D.; Varrone, S.; Gottesman, M.E.; Avvedimento, E.V.; Feliciello, A. Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J. Biol. Chem. 2003, 278, 4286–4294. [Google Scholar] [CrossRef]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.B.; Lendahl, U.; Zhao, J.; Nister, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, Y.; Hu, S.; Bi, S.; Li, S.; Hu, Y.; Kou, J.; Qi, J.; Yu, B. Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca(2+)-Calcineurin-Mediated Drp1 Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 1825. [Google Scholar] [CrossRef]
- Jahani-Asl, A.; Huang, E.; Irrcher, I.; Rashidian, J.; Ishihara, N.; Lagace, D.C.; Slack, R.S.; Park, D.S. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum. Mol. Genet. 2015, 24, 4573–4583. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Yan, J.; Liu, X.H.; Han, M.Z.; Wang, Y.M.; Sun, X.L.; Yu, N.; Li, T.; Su, B.; Chen, Z.Y. Blockage of GSK3beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Aging. 2015, 36, 211–227. [Google Scholar] [CrossRef]
- Gui, C.; Ren, Y.; Chen, J.; Wu, X.; Mao, K.; Li, H.; Yu, H.; Zou, F.; Li, W. p38 MAPK-DRP1 signaling is involved in mitochondrial dysfunction and cell death in mutant A53T alpha-synuclein model of Parkinson’s disease. Toxicol. Appl. Pharmacol. 2020, 388, 114874. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Smith, H.L.; Chau, K.Y.; Schapira, A.H. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Bruckner, A.; Kay, L.; Polge, C.; Epand, R.F.; Lee, R.M.; Lacombe, M.L.; Epand, R.M. Mitochondrial kinases and their molecular interaction with cardiolipin. Biochim. Biophys. Acta 2009, 1788, 2032–2047. [Google Scholar] [CrossRef] [PubMed]
- Cotteret, S.; Chernoff, J. Nucleocytoplasmic shuttling of Pak5 regulates its antiapoptotic properties. Mol. Cell Biol. 2006, 26, 3215–3230. [Google Scholar] [CrossRef][Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Murata, H.; Sakaguchi, M.; Kataoka, K.; Watanabe, M.; Nasu, Y.; Kumon, H.; Huh, N.H. Partial sensitization of human bladder cancer cells to a gene-therapeutic adenovirus carrying REIC/Dkk-3 by downregulation of BRPK/PINK1. Oncol. Rep. 2012, 27, 695–699. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Voigt, A.; Berlemann, L.A.; Winklhofer, K.F. The mitochondrial kinase PINK1: Functions beyond mitophagy. J. Neurochem. 2016, 139 (Suppl. 1), 232–239. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Ishihara-Paul, L.; Hulihan, M.M.; Kachergus, J.; Upmanyu, R.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Soto, A.; Kefi, M.; et al. PINK1 mutations and parkinsonism. Neurology 2008, 71, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, H. Parkin implicated in sporadic Parkinson’s disease. Lancet Neurol. 2005, 4, 798. [Google Scholar] [CrossRef]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Angeles, D.C.; Ho, P.; Chua, L.L.; Wang, C.; Yap, Y.W.; Ng, C.; Zhou, Z.; Lim, K.L.; Wszolek, Z.K.; Wang, H.Y.; et al. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum. Mol. Genet. 2014, 23, 3157–3165. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, K.H.; Cao, W.; Zeng, J.; Liao, H.; Zhao, L.; Guo, X. Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis 2010, 211, 216–223. [Google Scholar] [CrossRef]
- de la Cruz Lopez, K.G.; Toledo Guzman, M.E.; Sanchez, E.O.; Garcia Carranca, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef]
- Desai, B.N.; Myers, B.R.; Schreiber, S.L. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2002, 99, 4319–4324. [Google Scholar] [CrossRef]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.F.; Balaban, R.S.; Finkel, T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef]
- Lu, C.L.; Qin, L.; Liu, H.C.; Candas, D.; Fan, M.; Li, J.J. Tumor cells switch to mitochondrial oxidative phosphorylation under radiation via mTOR-mediated hexokinase II inhibition--a Warburg-reversing effect. PLoS ONE 2015, 10, e0121046. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Cotteret, S.; Jaffer, Z.M.; Beeser, A.; Chernoff, J. p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol. Cell Biol. 2003, 23, 5526–5539. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, D.; Bai, X.; Zou, H.; Lai, Y.; Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem. 2010, 285, 8621–8627. [Google Scholar] [CrossRef] [PubMed]
- Tasken, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Mishra, N.C.; Yoshida, K.; Kharbanda, S.; Saxena, S.; Kufe, D. Mitochondrial targeting of JNK/SAPK in the phorbol ester response of myeloid leukemia cells. Cell Death Differ. 2001, 8, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef]
- Ballard-Croft, C.; Kristo, G.; Yoshimura, Y.; Reid, E.; Keith, B.J.; Mentzer, R.M., Jr.; Lasley, R.D. Acute adenosine preconditioning is mediated by p38 MAPK activation in discrete subcellular compartments. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1359–H1366. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Shao, Y.; May, J.; Prou, D.; Perier, C.; Dauer, W.; Schon, E.A.; Przedborski, S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. USA 2008, 105, 12022–12027. [Google Scholar] [CrossRef]
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.W.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006, 60, 557–569. [Google Scholar] [CrossRef]
- Liang, J.; Xu, Z.X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.Y.; Peng, G.; Gambello, M.J.; et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015, 6, 7926. [Google Scholar] [CrossRef]
- Feng, Y.; Ariza, M.E.; Goulet, A.C.; Shi, J.; Nelson, M.A. Death-signal-induced relocalization of cyclin-dependent kinase 11 to mitochondria. Biochem. J. 2005, 392, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Bordin, L.; Cattapan, F.; Clari, G.; Toninello, A.; Siliprandi, N.; Moret, V. Spermine-mediated casein kinase II-uptake by rat liver mitochondria. Biochim. Biophys. Acta 1994, 1199, 266–270. [Google Scholar] [CrossRef]
- Clari, G.; Toninello, A.; Bordin, L.; Cattapan, F.; Piccinelli-Siliprandi, D.; Moret, V. Spermine effect on the binding of casein kinase I to the rat liver mitochondrial structures. Biochem. Biophys. Res. Commun. 1994, 205, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Singh, N.; Lawana, V.; Ghosh, A.; Harischandra, D.S.; Jin, H.; Hogan, C.; Sarkar, S.; Rokad, D.; Panicker, N.; et al. Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol. Dis. 2016, 93, 96–114. [Google Scholar] [CrossRef]
- Nowak, G.; Bakajsova, D. Protein kinase C-alpha interaction with F0F1-ATPase promotes F0F1-ATPase activity and reduces energy deficits in injured renal cells. J. Biol. Chem. 2015, 290, 7054–7066. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.M.; Li, X.Q.; Wang, Q.S.; Liu, Q.H.; Ginnan, R.; Singer, H.A.; Ho, Y.S.; Wang, Y.X. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 784–790. [Google Scholar] [CrossRef]
- Rubio, M.A.; Hopper, A.K. Transfer RNA travels from the cytoplasm to organelles. Wiley Interdiscip Rev. RNA 2011, 2, 802–817. [Google Scholar] [CrossRef]
- Sieber, F.; Duchene, A.M.; Marechal-Drouard, L. Mitochondrial RNA import: From diversity of natural mechanisms to potential applications. Int. Rev. Cell Mol. Biol. 2011, 287, 145–190. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Law, Y.S.; Ngan, L.; Yan, J.; Kwok, L.Y.; Sun, Y.; Cheng, S.; Schwenkert, S.; Lim, B.L. Multiple Kinases Can Phosphorylate the N-Terminal Sequences of Mitochondrial Proteins in Arabidopsis thaliana. Front. Plant. Sci. 2018, 9, 982. [Google Scholar] [CrossRef]
- Moulin, C.; Caumont-Sarcos, A.; Ieva, R. Mitochondrial presequence import: Multiple regulatory knobs fine-tune mitochondrial biogenesis and homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Vogtle, F.N.; Stojanovski, D.; Meisinger, C. Sorting and assembly of mitochondrial outer membrane proteins. Biochim. Biophys. Acta 2008, 1777, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Heazlewood, J.L.; Durek, P.; Hummel, J.; Selbig, J.; Weckwerth, W.; Walther, D.; Schulze, W.X. PhosPhAt: A database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 2008, 36, D1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Schmidt, O.; Harbauer, A.B.; Schonfisch, B.; Guiard, B.; Pfanner, N.; Meisinger, C. Biogenesis of the preprotein translocase of the outer mitochondrial membrane: Protein kinase A phosphorylates the precursor of Tom40 and impairs its import. Mol. Biol. Cell 2012, 23, 1618–1627. [Google Scholar] [CrossRef]
- Schmidt, O.; Harbauer, A.B.; Rao, S.; Eyrich, B.; Zahedi, R.P.; Stojanovski, D.; Schonfisch, B.; Guiard, B.; Sickmann, A.; Pfanner, N.; et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell 2011, 144, 227–239. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Gerbeth, C.; Schmidt, O.; Rao, S.; Harbauer, A.B.; Mikropoulou, D.; Opalinska, M.; Guiard, B.; Pfanner, N.; Meisinger, C. Glucose-induced regulation of protein import receptor Tom22 by cytosolic and mitochondria-bound kinases. Cell Metab 2013, 18, 578–587. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Becker, T.; Guiard, B.; Thornton, N.; Zufall, N.; Stroud, D.A.; Wiedemann, N.; Pfanner, N. Assembly of the mitochondrial protein import channel: Role of Tom5 in two-stage interaction of Tom40 with the SAM complex. Mol. Biol. Cell 2010, 21, 3106–3113. [Google Scholar] [CrossRef]
- Zaman, S.; Lippman, S.I.; Zhao, X.; Broach, J.R. How Saccharomyces responds to nutrients. Annu. Rev. Genet. 2008, 42, 27–81. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Pfannschmidt, S.; Guiard, B.; Stojanovski, D.; Milenkovic, D.; Kutik, S.; Pfanner, N.; Meisinger, C.; Wiedemann, N. Biogenesis of the mitochondrial TOM complex: Mim1 promotes insertion and assembly of signal-anchored receptors. J. Biol. Chem. 2008, 283, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Wenz, L.S.; Kruger, V.; Lehmann, W.; Muller, J.M.; Goroncy, L.; Zufall, N.; Lithgow, T.; Guiard, B.; Chacinska, A.; et al. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 2011, 194, 387–395. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Gerber, S.A. Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: Application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal. Chem. 2011, 83, 7635–7644. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef]
- Tsai, C.F.; Wang, Y.T.; Yen, H.Y.; Tsou, C.C.; Ku, W.C.; Lin, P.Y.; Chen, H.Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 2008, 104, 325–335. [Google Scholar] [CrossRef]
- Seifert, F.; Ciszak, E.; Korotchkina, L.; Golbik, R.; Spinka, M.; Dominiak, P.; Sidhu, S.; Brauer, J.; Patel, M.S.; Tittmann, K. Phosphorylation of serine 264 impedes active site accessibility in the E1 component of the human pyruvate dehydrogenase multienzyme complex. Biochemistry 2007, 46, 6277–6287. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab 2003, 284, E855–862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; McCarthy, D.B.; O’Connor, C.M.; Reed, L.J.; Stoops, J.K. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. USA 2001, 98, 14802–14807. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.C.; Pettit, F.H.; Reed, L.J. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 1969, 62, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Bowker-Kinley, M.; Popov, K.M. Evidence that pyruvate dehydrogenase kinase belongs to the ATPase/kinase superfamily. Biochem. J. 1999, 344 Pt. 1, 47–53. [Google Scholar] [CrossRef]
- Bowker-Kinley, M.M.; Davis, W.I.; Wu, P.; Harris, R.A.; Popov, K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329 Pt. 1, 191–196. [Google Scholar] [CrossRef]
- Steussy, C.N.; Popov, K.M.; Bowker-Kinley, M.M.; Sloan, R.B., Jr.; Harris, R.A.; Hamilton, J.A. Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J. Biol Chem 2001, 276, 37443–37450. [Google Scholar] [CrossRef]
- Teague, W.M.; Pettit, F.H.; Yeaman, S.J.; Reed, L.J. Function of phosphorylation sites on pyruvate dehydrogenase. Biochem. Biophys. Res. Commun. 1979, 87, 244–252. [Google Scholar] [CrossRef]
- Korotchkina, L.G.; Patel, M.S. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 2001, 276, 37223–37229. [Google Scholar] [CrossRef]
- Korotchkina, L.G.; Patel, M.S. Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J. Biol. Chem. 2001, 276, 5731–5738. [Google Scholar] [CrossRef]
- Kolobova, E.; Tuganova, A.; Boulatnikov, I.; Popov, K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 2001, 358, 69–77. [Google Scholar] [CrossRef]
- Kerbey, A.L.; Randle, P.J.; Cooper, R.H.; Whitehouse, S.; Pask, H.T.; Denton, R.M. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: Role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem. J. 1976, 154, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Sato, Y.; Oshida, Y.; Yoshimura, A.; Fujitsuka, N.; Sugiyama, S.; Shimomura, Y. Effects of aging on the activities of pyruvate dehydrogenase complex and its kinase in rat heart. Life Sci. 1997, 60, 2309–2314. [Google Scholar] [CrossRef]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. Activation of c-Jun-N-terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 2009, 583, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Andersen, J.K. The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 2003, 55, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Yang, W.; Lu, Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett. 2013, 339, 153–158. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Pyruvate kinase M2 at a glance. J. Cell Sci. 2015, 128, 1655–1660. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Hol, W.G. Crystal structures of substrates and products bound to the phosphoglycerate kinase active site reveal the catalytic mechanism. Biochemistry 1998, 37, 4429–4436. [Google Scholar] [CrossRef]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Chiu, L.L.; Sethi, S.K.; Koay, E.S. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell Proteom. 2005, 4, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Liang, Y.; Chien, K.Y.; Yu, J.S. Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics 2006, 6, 2259–2272. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Lamendola, D.E.; Yusuf, R.Z.; Penson, R.T.; Preffer, F.I.; Seiden, M.V. Overexpression of human phosphoglycerate kinase 1 (PGK1) induces a multidrug resistance phenotype. Anticancer Res. 2002, 22, 1933–1941. [Google Scholar] [PubMed]
- Ahmad, S.S.; Glatzle, J.; Bajaeifer, K.; Buhler, S.; Lehmann, T.; Konigsrainer, I.; Vollmer, J.P.; Sipos, B.; Ahmad, S.S.; Northoff, H.; et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 2013, 43, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Huang, H.; Lv, X.; Tang, Z.; Chen, M.; Chen, T.; Duan, W.; Sun, H.; Li, Q.; Tan, R.; et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell Physiol. Biochem. 2011, 27, 207–216. [Google Scholar] [CrossRef]
- Zieker, D.; Konigsrainer, I.; Tritschler, I.; Loffler, M.; Beckert, S.; Traub, F.; Nieselt, K.; Buhler, S.; Weller, M.; Gaedcke, J.; et al. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int. J. Cancer 2010, 126, 1513–1520. [Google Scholar] [CrossRef]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef]
- Song, J.S.; Yang, S.D. Tau protein kinase I/GSK-3 beta/kinase FA in heparin phosphorylates tau on Ser199, Thr231, Ser235, Ser262, Ser369, and Ser400 sites phosphorylated in Alzheimer disease brain. J. Protein Chem. 1995, 14, 95–105. [Google Scholar] [CrossRef]
- Bykova, N.V.; Stensballe, A.; Egsgaard, H.; Jensen, O.N.; Moller, I.M. Phosphorylation of formate dehydrogenase in potato tuber mitochondria. J. Biol. Chem. 2003, 278, 26021–26030. [Google Scholar] [CrossRef]
- Chen, X.J.; Butow, R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef]
- Shadel, G.S. Mitochondrial DNA, aconitase ‘wraps’ it up. Trends Biochem. Sci. 2005, 30, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Lewandrowski, U.; Sickmann, A.; Cesaro, L.; Brunati, A.M.; Toninello, A.; Salvi, M. Identification of new tyrosine phosphorylated proteins in rat brain mitochondria. FEBS Lett. 2008, 582, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Niemi, N.M.; Hutchins, P.D.; Condon, S.G.F.; Jochem, A.; Ulbrich, A.; Higbee, A.J.; Russell, J.D.; Senes, A.; Coon, J.J.; et al. Ptc7p Dephosphorylates Select Mitochondrial Proteins to Enhance Metabolic Function. Cell Rep. 2017, 18, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.; Wenz, T. Post-translational modification of mitochondria as a novel mode of regulation. Exp. Gerontol. 2014, 56, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Castillo, A.F.; Podesta, E.J.; Poderoso, C. Mitochondrial fusion and ERK activity regulate steroidogenic acute regulatory protein localization in mitochondria. PLoS ONE 2014, 9, e100387. [Google Scholar] [CrossRef] [PubMed]
- Bose, H.S.; Lingappa, V.R.; Miller, W.L. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 2002, 417, 87–91. [Google Scholar] [CrossRef]
- Castillo, A.F.; Orlando, U.; Helfenberger, K.E.; Poderoso, C.; Podesta, E.J. The role of mitochondrial fusion and StAR phosphorylation in the regulation of StAR activity and steroidogenesis. Mol. Cell Endocrinol. 2015, 408, 73–79. [Google Scholar] [CrossRef]
- Arakane, F.; King, S.R.; Du, Y.; Kallen, C.B.; Walsh, L.P.; Watari, H.; Stocco, D.M.; Strauss, J.F., 3rd. Phosphorylation of steroidogenic acute regulatory protein (StAR) modulates its steroidogenic activity. J. Biol. Chem. 1997, 272, 32656–32662. [Google Scholar] [CrossRef]
- Fleury, A.; Mathieu, A.P.; Ducharme, L.; Hales, D.B.; LeHoux, J.G. Phosphorylation and function of the hamster adrenal steroidogenic acute regulatory protein (StAR). J. Steroid Biochem. Mol. Biol. 2004, 91, 259–271. [Google Scholar] [CrossRef]
- Granot, Z.; Kobiler, O.; Melamed-Book, N.; Eimerl, S.; Bahat, A.; Lu, B.; Braun, S.; Maurizi, M.R.; Suzuki, C.K.; Oppenheim, A.B.; et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: The unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007, 21, 2164–2177. [Google Scholar] [CrossRef]
- van den Heuvel, L.; Smeitink, J. The oxidative phosphorylation (OXPHOS) system: Nuclear genes and human genetic diseases. Bioessays 2001, 23, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Nosek, J.; Fukuhara, H. NADH dehydrogenase subunit genes in the mitochondrial DNA of yeasts. J. Bacteriol. 1994, 176, 5622–5630. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fearnley, I.M.; Peak-Chew, S.Y.; Walker, J.E. The phosphorylation of subunits of complex I from bovine heart mitochondria. J. Biol. Chem. 2004, 279, 26036–26045. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; De Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian complex I: A regulable and vulnerable pacemaker in mitochondrial respiratory function. Biochim. Biophys. Acta 2008, 1777, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Gowthami, N.; Sunitha, B.; Kumar, M.; Keshava Prasad, T.S.; Gayathri, N.; Padmanabhan, B.; Srinivas Bharath, M.M. Mapping the protein phosphorylation sites in human mitochondrial complex I (NADH: Ubiquinone oxidoreductase): A bioinformatics study with implications for brain aging and neurodegeneration. J. Chem. Neuroanat 2019, 95, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Scacco, S.; Vergari, R.; Scarpulla, R.C.; Technikova-Dobrova, Z.; Sardanelli, A.; Lambo, R.; Lorusso, V.; Papa, S. cAMP-dependent phosphorylation of the nuclear encoded 18-kDa (IP) subunit of respiratory complex I and activation of the complex in serum-starved mouse fibroblast cultures. J. Biol. Chem. 2000, 275, 17578–17582. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation pattern of the NDUFS4 subunit of complex I of the mammalian respiratory chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef]
- Papa, S.; Sardanelli, A.M.; Cocco, T.; Speranza, F.; Scacco, S.C.; Technikova-Dobrova, Z. The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett. 1996, 379, 299–301. [Google Scholar] [CrossRef]
- Papa, S.; Scacco, S.; Sardanelli, A.M.; Vergari, R.; Papa, F.; Budde, S.; van den Heuvel, L.; Smeitink, J. Mutation in the NDUFS4 gene of complex I abolishes cAMP-dependent activation of the complex in a child with fatal neurological syndrome. FEBS Lett. 2001, 489, 259–262. [Google Scholar] [CrossRef]
- van den Heuvel, L.; Ruitenbeek, W.; Smeets, R.; Gelman-Kohan, Z.; Elpeleg, O.; Loeffen, J.; Trijbels, F.; Mariman, E.; de Bruijn, D.; Smeitink, J. Demonstration of a new pathogenic mutation in human complex I deficiency: A 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am. J. Hum. Genet. 1998, 62, 262–268. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Larizza, M.; Pacelli, C.; Cocco, T.; Papa, S. Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic. Biol. Med. 2012, 52, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grunewald, A.; Seibler, P.; et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, M.; Candas, D.; Zhang, T.Q.; Qin, L.; Eldridge, A.; Wachsmann-Hogiu, S.; Ahmed, K.M.; Chromy, B.A.; Nantajit, D.; et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev. Cell 2014, 29, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Inoue, T.; Yamaki, J.; Homma, M.K.; Kurosaki, T.; Homma, Y. Mitochondrial reactive oxygen species suppress humoral immune response through reduction of CD19 expression in B cells in mice. Eur. J. Immunol. 2017, 47, 406–418. [Google Scholar] [CrossRef]
- Zhao, X.; Leon, I.R.; Bak, S.; Mogensen, M.; Wrzesinski, K.; Hojlund, K.; Jensen, O.N. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol. Cell Proteom. 2011, 10, M110–000299. [Google Scholar] [CrossRef]
- Arachiche, A.; Augereau, O.; Decossas, M.; Pertuiset, C.; Gontier, E.; Letellier, T.; Dachary-Prigent, J. Localization of PTP-1B, SHP-2, and Src exclusively in rat brain mitochondria and functional consequences. J. Biol. Chem. 2008, 283, 24406–24411. [Google Scholar] [CrossRef]
- Mahapatra, G.; Varughese, A.; Ji, Q.; Lee, I.; Liu, J.; Vaishnav, A.; Sinkler, C.; Kapralov, A.A.; Moraes, C.T.; Sanderson, T.H.; et al. Phosphorylation of Cytochrome c Threonine 28 Regulates Electron Transport Chain Activity in Kidney: Implications for Amp Kinase. J. Biol. Chem. 2017, 292, 64–79. [Google Scholar] [CrossRef]
- Pecina, P.; Borisenko, G.G.; Belikova, N.A.; Tyurina, Y.Y.; Pecinova, A.; Lee, I.; Samhan-Arias, A.K.; Przyklenk, K.; Kagan, V.E.; Huttemann, M. Phosphomimetic substitution of cytochrome C tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 2010, 49, 6705–6714. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Kalpage, H.A.; Vaishnav, A.; Liu, J.; Lee, I.; Mahapatra, G.; Turner, A.A.; Zurek, M.P.; Ji, Q.; Moraes, C.T.; et al. Regulation of Respiration and Apoptosis by Cytochrome c Threonine 58 Phosphorylation. Sci. Rep. 2019, 9, 15815. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Beltran, B.; Guerra-Castellano, A.; Diaz-Quintana, A.; Del Conte, R.; Garcia-Maurino, S.M.; Diaz-Moreno, S.; Gonzalez-Arzola, K.; Santos-Ocana, C.; Velazquez-Campoy, A.; De la Rosa, M.A.; et al. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc. Natl. Acad. Sci. USA 2017, 114, E3041–E3050. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Lee, I.; Huttemann, M. Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling. Cells 2020, 9, 1843. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. Human cytochrome c oxidase: Structure, function, and deficiency. J. Bioenerg. Biomembr. 1997, 29, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Salomon, A.R.; Ficarro, S.; Mathes, I.; Lottspeich, F.; Grossman, L.I.; Huttemann, M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J. Biol. Chem. 2005, 280, 6094–6100. [Google Scholar] [CrossRef]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Huttemann, M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef]
- Prabu, S.K.; Anandatheerthavarada, H.K.; Raza, H.; Srinivasan, S.; Spear, J.F.; Avadhani, N.G. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem. 2006, 281, 2061–2070. [Google Scholar] [CrossRef]
- Srinivasan, S.; Spear, J.; Chandran, K.; Joseph, J.; Kalyanaraman, B.; Avadhani, N.G. Oxidative stress induced mitochondrial protein kinase A mediates cytochrome c oxidase dysfunction. PLoS ONE 2013, 8, e77129. [Google Scholar] [CrossRef]
- Kunova, N.; Ondrovicova, G.; Bauer, J.A.; Bellova, J.; Ambro, L.; Martinakova, L.; Kotrasova, V.; Kutejova, E.; Pevala, V. The role of Lon-mediated proteolysis in the dynamics of mitochondrial nucleic acid-protein complexes. Sci. Rep. 2017, 7, 631. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Angireddy, R.; Srinivasan, S.; Guha, M.; Spear, J.; Lu, B.; Anandatheerthavarada, H.K.; Suzuki, C.K.; Avadhani, N.G. Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: Coupled mechanisms of energy metabolism regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.; Lin, D.; Akoyev, V.; Willard, L.; Takemoto, D. Protein kinase C epsilon activates lens mitochondrial cytochrome c oxidase subunit IV during hypoxia. Exp. Eye Res. 2008, 86, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Ogbi, M.; Johnson, J.A. Protein kinase Cepsilon interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem. J. 2006, 393, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Struglics, A.; Fredlund, K.M.; Moller, I.M.; Allen, J.F. Two subunits of the F0F1-ATPase are phosphorylated in the inner mitochondrial membrane. Biochem. Biophys. Res. Commun. 1998, 243, 664–668. [Google Scholar] [CrossRef]
- Hojlund, K.; Wrzesinski, K.; Larsen, P.M.; Fey, S.J.; Roepstorff, P.; Handberg, A.; Dela, F.; Vinten, J.; McCormack, J.G.; Reynet, C.; et al. Proteome analysis reveals phosphorylation of ATP synthase beta -subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J. Biol. Chem. 2003, 278, 10436–10442. [Google Scholar] [CrossRef]
- Hojlund, K.; Yi, Z.; Lefort, N.; Langlais, P.; Bowen, B.; Levin, K.; Beck-Nielsen, H.; Mandarino, L.J. Human ATP synthase beta is phosphorylated at multiple sites and shows abnormal phosphorylation at specific sites in insulin-resistant muscle. Diabetologia 2010, 53, 541–551. [Google Scholar] [CrossRef]
- Yang, J.Y.; Deng, W.; Chen, Y.; Fan, W.; Baldwin, K.M.; Jope, R.S.; Wallace, D.C.; Wang, P.H. Impaired translocation and activation of mitochondrial Akt1 mitigated mitochondrial oxidative phosphorylation Complex V activity in diabetic myocardium. J. Mol. Cell Cardiol. 2013, 59, 167–175. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Sanchez-Arago, M.; Soldevilla, B.; Del Arco, A.; Nuevo-Tapioles, C.; Cuezva, J.M. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H(+)-ATP Synthase. Cell Rep. 2015, 12, 2143–2155. [Google Scholar] [CrossRef]
- Pullman, M.E.; Monroy, G.C. A Naturally Occurring Inhibitor of Mitochondrial Adenosine Triphosphatase. J. Biol. Chem. 1963, 238, 3762–3769. [Google Scholar] [CrossRef]
- Garcia-Aguilar, A.; Cuezva, J.M. A Review of the Inhibition of the Mitochondrial ATP Synthase by IF1 in vivo: Reprogramming Energy Metabolism and Inducing Mitohormesis. Front. Physiol. 2018, 9, 1322. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, E.; Lanning, N.J. Phosphorylation of OXPHOS Machinery Subunits: Functional Implications in Cell Biology and Disease. Yale J. Biol. Med. 2019, 92, 523–531. [Google Scholar] [PubMed]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. 1), S130–S135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, A.; Oh, K.J.; Lee, S.C.; Kim, W.K.; Bae, K.H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D. Uncoupling protein 1 of brown adipocytes, the only uncoupler: A historical perspective. Front. Endocrinol 2011, 2, 85. [Google Scholar] [CrossRef]
- Jezek, P.; Jaburek, M.; Porter, R.K. Uncoupling mechanism and redox regulation of mitochondrial uncoupling protein 1 (UCP1). Biochim. Biophys. Acta Bioenerg. 2019, 1860, 259–269. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Locke, R.M. Thermogenic mechanisms in brown fat. Physiol. Rev. 1984, 64, 1–64. [Google Scholar] [CrossRef]
- Macher, G.; Koehler, M.; Rupprecht, A.; Kreiter, J.; Hinterdorfer, P.; Pohl, E.E. Inhibition of mitochondrial UCP1 and UCP3 by purine nucleotides and phosphate. Biochim. Biophys. Acta Biomembr. 2018, 1860, 664–672. [Google Scholar] [CrossRef]
- Bast-Habersbrunner, A.; Fromme, T. Purine Nucleotides in the Regulation of Brown Adipose Tissue Activity. Front. Endocrinol. 2020, 11, 118. [Google Scholar] [CrossRef]
- Villarroya, F.; Peyrou, M.; Giralt, M. Transcriptional regulation of the uncoupling protein-1 gene. Biochimie 2017, 134, 86–92. [Google Scholar] [CrossRef]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Ohyama, K.; Hasegawa, Y.; Chang, H.Y.; Ogura, M.; Sato, A.; Hong, H.; Hosono, T.; Sharp, L.Z.; Scheel, D.W.; et al. Phosphoproteomics Identifies CK2 as a Negative Regulator of Beige Adipocyte Thermogenesis and Energy Expenditure. Cell Metab. 2015, 22, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.M.; Porter, R.K.; Morrice, N.A. Identification of serine phosphorylation in mitochondrial uncoupling protein 1. Biochim. Biophys. Acta 2008, 1777, 1060–1065. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anand, R.; Langer, T.; Baker, M.J. Proteolytic control of mitochondrial function and morphogenesis. Biochim. Biophys. Acta 2013, 1833, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect Biol. 2011, 3. [Google Scholar] [CrossRef]
- Bender, T.; Lewrenz, I.; Franken, S.; Baitzel, C.; Voos, W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 2011, 22, 541–554. [Google Scholar] [CrossRef]
- Lund, A.A.; Rhoads, D.M.; Lund, A.L.; Cerny, R.L.; Elthon, T.E. In vivo modifications of the maize mitochondrial small heat stress protein, HSP22. J Biol. Chem. 2001, 276, 29924–29929. [Google Scholar] [CrossRef]
- Downs, C.A.; Heckathorn, S.A. The mitochondrial small heat-shock protein protects NADH:ubiquinone oxidoreductase of the electron transport chain during heat stress in plants. FEBS Lett. 1998, 430, 246–250. [Google Scholar] [CrossRef]
- Lund, A.A.; Blum, P.H.; Bhattramakki, D.; Elthon, T.E. Heat-stress response of maize mitochondria. Plant. Physiol. 1998, 116, 1097–1110. [Google Scholar] [CrossRef]
- Lenne, C.; Douce, R. A Low Molecular Mass Heat-Shock Protein Is Localized to Higher Plant Mitochondria. Plant. Physiol. 1994, 105, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Chen, C.Y.; Lin, T.H.; Huang, Z.W.; Chi, T.H.; Ma, Y.S.; Wu, S.B.; Wei, Y.H.; Hsieh, M. The protective roles of phosphorylated heat shock protein 27 in human cells harboring myoclonus epilepsy with ragged-red fibers A8344G mtDNA mutation. FEBS J. 2012, 279, 2987–3001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Lambert, H.; Landry, J. Transient activation of a distinct serine protein kinase is responsible for 27-kDa heat shock protein phosphorylation in mitogen-stimulated and heat-shocked cells. J. Biol. Chem. 1993, 268, 35–43. [Google Scholar] [CrossRef]
- Arrigo, A.P. Human small heat shock proteins: Protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 2013, 587, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Hadari, Y.R.; Haring, H.U.; Zick, Y. p75, a member of the heat shock protein family, undergoes tyrosine phosphorylation in response to oxidative stress. J. Biol. Chem. 1997, 272, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab 2013, 17, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 as a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ficarro, S.; Chertihin, O.; Westbrook, V.A.; White, F.; Jayes, F.; Kalab, P.; Marto, J.A.; Shabanowitz, J.; Herr, J.C.; Hunt, D.F.; et al. Phosphoproteome analysis of capacitated human sperm. Evidence of tyrosine phosphorylation of a kinase-anchoring protein 3 and valosin-containing protein/p97 during capacitation. J. Biol. Chem. 2003, 278, 11579–11589. [Google Scholar] [CrossRef]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Landry, S.J.; Georgopoulos, C. The ins and outs of a molecular chaperone machine. Trends Biochem. Sci. 1998, 23, 138–143. [Google Scholar] [CrossRef]
- Vilasi, S.; Bulone, D.; Caruso Bavisotto, C.; Campanella, C.; Marino Gammazza, A.; San Biagio, P.L.; Cappello, F.; Conway de Macario, E.; Macario, A.J.L. Chaperonin of Group I: Oligomeric Spectrum and Biochemical and Biological Implications. Front. Mol. Biosci. 2017, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Caruso Bavisotto, C.; Alberti, G.; Vitale, A.M.; Paladino, L.; Campanella, C.; Rappa, F.; Gorska, M.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; et al. Hsp60 Post-translational Modifications: Functional and Pathological Consequences. Front. Mol. Biosci. 2020, 7, 95. [Google Scholar] [CrossRef]
- Gu, Y.; Ande, S.R.; Mishra, S. Altered O-GlcNAc modification and phosphorylation of mitochondrial proteins in myoblast cells exposed to high glucose. Arch. Biochem. Biophys. 2011, 505, 98–104. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Mukherjee, A.; Patra, U.; Bhowmick, R.; Basak, T.; Sengupta, S.; Chawla-Sarkar, M. Tyrosine phosphorylation modulates mitochondrial chaperonin Hsp60 and delays rotavirus NSP4-mediated apoptotic signaling in host cells. Cell Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Asquith, K.L.; Baleato, R.M.; McLaughlin, E.A.; Nixon, B.; Aitken, R.J. Tyrosine phosphorylation activates surface chaperones facilitating sperm-zona recognition. J. Cell Sci. 2004, 117, 3645–3657. [Google Scholar] [CrossRef] [PubMed]
- Desautels, M.; Goldberg, A.L. Liver mitochondria contain an ATP-dependent, vanadate-sensitive pathway for the degradation of proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 1869–1873. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 2019, 38, 6926–6939. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef]
- Gibellini, L.; De Gaetano, A.; Mandrioli, M.; Van Tongeren, E.; Bortolotti, C.A.; Cossarizza, A.; Pinti, M. The biology of Lonp1: More than a mitochondrial protease. Int. Rev. Cell Mol. Biol. 2020, 354, 1–61. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Tammineni, P.; Mohammed, F.; Paripati, A. Nuclear Transcription Factors in the Mitochondria: A New Paradigm in Fine-Tuning Mitochondrial Metabolism. Handb. Exp. Pharmacol. 2017, 240, 3–20. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Goto, M.; Miwa, H.; Suganuma, K.; Tsunekawa-Imai, N.; Shikami, M.; Mizutani, M.; Mizuno, S.; Hanamura, I.; Nitta, M. Adaptation of leukemia cells to hypoxic condition through switching the energy metabolism or avoiding the oxidative stress. BMC Cancer 2014, 14, 76. [Google Scholar] [CrossRef]
- Zhou, X.; Teper, D.; Andrade, M.O.; Zhang, T.; Chen, S.; Song, W.Y.; Wang, N. A Phosphorylation Switch on Lon Protease Regulates Bacterial Type III Secretion System in Host. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Brunings, A.M.; Gabriel, D.W. Xanthomonas citri: Breaking the surface. Mol. Plant. Pathol. 2003, 4, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Ortega, J.; Singh, S.K.; Wang, N.; Huang, N.N.; Steven, A.C.; Maurizi, M.R. Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J. Biol. Chem. 2002, 277, 21095–21102. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, P.C.; He, J.; Morris, E.P. Molecular model of the human 26S proteasome. Mol. Cell 2012, 46, 54–66. [Google Scholar] [CrossRef]
- Kang, S.G.; Maurizi, M.R.; Thompson, M.; Mueser, T.; Ahvazi, B. Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J. Struct. Biol. 2004, 148, 338–352. [Google Scholar] [CrossRef]
- Kang, S.G.; Dimitrova, M.N.; Ortega, J.; Ginsburg, A.; Maurizi, M.R. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 2005, 280, 35424–35432. [Google Scholar] [CrossRef]
- Kasashima, K.; Sumitani, M.; Endo, H. Maintenance of mitochondrial genome distribution by mitochondrial AAA+ protein ClpX. Exp. Cell Res. 2012, 318, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Bolon, D.N.; Burton, B.M.; Burton, R.E.; Flynn, J.M.; Grant, R.A.; Hersch, G.L.; Joshi, S.A.; Kenniston, J.A.; Levchenko, I.; et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 2004, 119, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Dohi, T.; Raskett, C.M.; Orlowski, G.M.; Powers, C.M.; Gilbert, C.A.; Ross, A.H.; Plescia, J.; Altieri, D.C. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Invest. 2011, 121, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Brown, T.A.; Tkachuk, A.N.; Shtengel, G.; Kopek, B.G.; Bogenhagen, D.F.; Hess, H.F.; Clayton, D.A. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell Biol. 2011, 31, 4994–5010. [Google Scholar] [CrossRef]
- Vozarikova, V.; Kunova, N.; Bauer, J.A.; Frankovsky, J.; Kotrasova, V.; Prochazkova, K.; Dzugasova, V.; Kutejova, E.; Pevala, V.; Nosek, J.; et al. Mitochondrial HMG-Box Containing Proteins: From Biochemical Properties to the Roles in Human Diseases. Biomolecules 2020, 10, 1193. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lee, Y.K.; Chae, C.B. The modulation of the biological activities of mitochondrial histone Abf2p by yeast PKA and its possible role in the regulation of mitochondrial DNA content during glucose repression. Biochim. Biophys. Acta 2001, 1522, 175–186. [Google Scholar] [CrossRef]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Z.; Zhu, J.; Dagda, R.K.; Uechi, G.; Cherra, S.J., 3rd; Gusdon, A.M.; Balasubramani, M.; Chu, C.T. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: Implications for Parkinson’s disease. Mitochondrion 2014, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Alami-Ouahabi, N.; Veilleux, S.; Meistrich, M.L.; Boissonneault, G. The testis-specific high-mobility-group protein, a phosphorylation-dependent DNA-packaging factor of elongating and condensing spermatids. Mol. Cell Biol. 1996, 16, 3720–3729. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, C.; Yau, P.; Bradbury, E.M.; Shyamala, G.; Yasuda, H.; Walsh, D.A. Phosphorylation of high-mobility-group proteins by the calcium-phospholipid-dependent protein kinase and the cyclic AMP-dependent protein kinase. J. Biol. Chem. 1984, 259, 13495–13503. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Schulze, E. High affinity interaction of dipteran high mobility group (HMG) proteins 1 with DNA is modulated by COOH-terminal regions flanking the HMG box domain. J. Biol. Chem. 1994, 269, 10713–10719. [Google Scholar] [CrossRef]
- Lund, T.; Berg, K. Metaphase-specific phosphorylations weaken the association between chromosomal proteins HMG 14 and 17, and DNA. FEBS Lett. 1991, 289, 113–116. [Google Scholar] [CrossRef]
- Reeves, R.; Langan, T.A.; Nissen, M.S. Phosphorylation of the DNA-binding domain of nonhistone high-mobility group I protein by cdc2 kinase: Reduction of binding affinity. Proc. Natl. Acad. Sci. USA 1991, 88, 1671–1675. [Google Scholar] [CrossRef]
- Schwanbeck, R.; Wisniewski, J.R. Cdc2 and mitogen-activated protein kinases modulate DNA binding properties of the putative transcriptional regulator Chironomus high mobility group protein I. J. Biol. Chem. 1997, 272, 27476–27483. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cosials, A.; Sidow, J.F.; Jimenez-Menendez, N.; Fernandez-Millan, P.; Montoya, J.; Jacobs, H.T.; Coll, M.; Bernado, P.; Sola, M. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nat. Struct. Mol. Biol. 2011, 18, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, M.; Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. The mitochondrial RNA polymerase contributes critically to promoter specificity in mammalian cells. EMBO J. 2004, 23, 4606–4614. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: Implications for Parkinson’s disease. Autophagy 2008, 4, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Marko, A.J.; Miller, R.A.; Kelman, A.; Frauwirth, K.A. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS ONE 2010, 5, e15425. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Rose, A.; Meier, I. MFP1 is a thylakoid-associated, nucleoid-binding protein with a coiled-coil structure. Nucleic Acids Res. 2003, 31, 5175–5185. [Google Scholar] [CrossRef] [PubMed]
- Ogrzewalla, K.; Piotrowski, M.; Reinbothe, S.; Link, G. The plastid transcription kinase from mustard (Sinapis alba L.). A nuclear-encoded CK2-type chloroplast enzyme with redox-sensitive function. Eur. J. Biochem. 2002, 269, 3329–3337. [Google Scholar] [CrossRef]
- Powikrowska, M.; Oetke, S.; Jensen, P.E.; Krupinska, K. Dynamic composition, shaping and organization of plastid nucleoids. Front. Plant. Sci. 2014, 5, 424. [Google Scholar] [CrossRef]
- Sekine, K.; Fujiwara, M.; Nakayama, M.; Takao, T.; Hase, T.; Sato, N. DNA binding and partial nucleoid localization of the chloroplast stromal enzyme ferredoxin:sulfite reductase. FEBS J. 2007, 274, 2054–2069. [Google Scholar] [CrossRef]
- Sekine, K.; Hase, T.; Sato, N. Reversible DNA compaction by sulfite reductase regulates transcriptional activity of chloroplast nucleoids. J. Biol. Chem. 2002, 277, 24399–24404. [Google Scholar] [CrossRef]
- Melonek, J.; Matros, A.; Trosch, M.; Mock, H.P.; Krupinska, K. The core of chloroplast nucleoids contains architectural SWIB domain proteins. Plant. Cell 2012, 24, 3060–3073. [Google Scholar] [CrossRef] [PubMed]
- Chi-Ham, C.L.; Keaton, M.A.; Cannon, G.C.; Heinhorst, S. The DNA-compacting protein DCP68 from soybean chloroplasts is ferredoxin:sulfite reductase and co-localizes with the organellar nucleoid. Plant. Mol. Biol. 2002, 49, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Tomaska, L. Phosphorylation of mitochondrial telomere binding protein of Candida parapsilosis by camp-dependent protein kinase. Biochem. Biophys. Res. Commun. 1998, 242, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, M.; Jaehning, J.A. A mutation in the yeast mitochondrial core RNA polymerase, Rpo41, confers defects in both specificity factor interaction and promoter utilization. J. Biol. Chem. 2004, 279, 2012–2019. [Google Scholar] [CrossRef]
- Cotney, J.; Shadel, G.S. Evidence for an early gene duplication event in the evolution of the mitochondrial transcription factor B family and maintenance of rRNA methyltransferase activity in human mtTFB1 and mtTFB2. J. Mol. Evol. 2006, 63, 707–717. [Google Scholar] [CrossRef]
- Shutt, T.E.; Gray, M.W. Homologs of mitochondrial transcription factor B, sparsely distributed within the eukaryotic radiation, are likely derived from the dimethyladenosine methyltransferase of the mitochondrial endosymbiont. Mol. Biol. Evol. 2006, 23, 1169–1179. [Google Scholar] [CrossRef]
- Gnad, F.; de Godoy, L.M.; Cox, J.; Neuhauser, N.; Ren, S.; Olsen, J.V.; Mann, M. High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 2009, 9, 4642–4652. [Google Scholar] [CrossRef]
- Soufi, B.; Kelstrup, C.D.; Stoehr, G.; Frohlich, F.; Walther, T.C.; Olsen, J.V. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol. Biosyst. 2009, 5, 1337–1346. [Google Scholar] [CrossRef]
- Prieto-Martin, A.; Montoya, J.; Martinez-Azorin, F. Phosphorylation of rat mitochondrial transcription termination factor (mTERF) is required for transcription termination but not for binding to DNA. Nucleic Acids Res. 2004, 32, 2059–2068. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Silva, P.; Martinez-Azorin, F.; Micol, V.; Attardi, G. The human mitochondrial transcription termination factor (mTERF) is a multizipper protein but binds to DNA as a monomer, with evidence pointing to intramolecular leucine zipper interactions. EMBO J. 1997, 16, 1066–1079. [Google Scholar] [CrossRef]
- Cammarota, M.; Paratcha, G.; Bevilaqua, L.R.; Levi de Stein, M.; Lopez, M.; Pellegrino de Iraldi, A.; Izquierdo, I.; Medina, J.H. Cyclic AMP-responsive element binding protein in brain mitochondria. J. Neurochem. 1999, 72, 2272–2277. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, C.H.; Simon, D.K.; Aminova, L.R.; Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Lonze, B.E.; Kim, K.S.; Ginty, D.D.; et al. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J. Biol. Chem. 2005, 280, 40398–40401. [Google Scholar] [CrossRef] [PubMed]
- Marinov, G.K.; Wang, Y.E.; Chan, D.; Wold, B.J. Evidence for site-specific occupancy of the mitochondrial genome by nuclear transcription factors. PLoS ONE 2014, 9, e84713. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zassenhaus, H.P. Purification and characterization of an RNA dodecamer sequence binding protein from mitochondria of Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1999, 261, 740–745. [Google Scholar] [CrossRef]
- Li, H.; Zassenhaus, H.P. Phosphorylation is required for high-affinity binding of DBP, a yeast mitochondrial site-specific RNA binding protein. Curr. Genet. 2000, 37, 356–363. [Google Scholar] [CrossRef]
- Dziembowski, A.; Piwowarski, J.; Hoser, R.; Minczuk, M.; Dmochowska, A.; Siep, M.; van der Spek, H.; Grivell, L.; Stepien, P.P. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J. Biol. Chem. 2003, 278, 1603–1611. [Google Scholar] [CrossRef]
- Hofmann, T.J.; Min, J.; Zassenhaus, H.P. Formation of the 3’ end of yeast mitochondrial mRNAs occurs by site-specific cleavage two bases downstream of a conserved dodecamer sequence. Yeast 1993, 9, 1319–1330. [Google Scholar] [CrossRef]
- Osinga, K.A.; De Vries, E.; Van der Horst, G.; Tabak, H.F. Processing of yeast mitochondrial messenger RNAs at a conserved dodecamer sequence. EMBO J. 1984, 3, 829–834. [Google Scholar] [CrossRef]
- He, H.; Chen, M.; Scheffler, N.K.; Gibson, B.W.; Spremulli, L.L.; Gottlieb, R.A. Phosphorylation of mitochondrial elongation factor Tu in ischemic myocardium: Basis for chloramphenicol-mediated cardioprotection. Circ. Res. 2001, 89, 461–467. [Google Scholar] [CrossRef]
- Lippmann, C.; Lindschau, C.; Vijgenboom, E.; Schroder, W.; Bosch, L.; Erdmann, V.A. Prokaryotic elongation factor Tu is phosphorylated in vivo. J. Biol. Chem. 1993, 268, 601–607. [Google Scholar] [CrossRef]
- He, H.; Li, H.L.; Lin, A.; Gottlieb, R.A. Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia. Cell Death Differ. 1999, 6, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5’ -> 3’ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Tatarov, O.; Mitchell, T.J.; Seywright, M.; Leung, H.Y.; Brunton, V.G.; Edwards, J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin. Cancer Res. 2009, 15, 3540–3549. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, B.S.; Vroom, T.M.; Adriaansen-Slot, S.S.; Ottenhoff-Kalff, A.E.; Geertzema, J.G.; Hennipman, A.; Rijksen, G. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J. Pathol. 1996, 180, 383–388. [Google Scholar] [CrossRef]
- Fu, Y.; Zagozdzon, R.; Avraham, R.; Avraham, H.K. CHK negatively regulates Lyn kinase and suppresses pancreatic cancer cell invasion. Int. J. Oncol. 2006, 29, 1453–1458. [Google Scholar] [CrossRef]
- Bolen, J.B.; Veillette, A.; Schwartz, A.M.; Deseau, V.; Rosen, N. Analysis of pp60c-src in human colon carcinoma and normal human colon mucosal cells. Oncogene Res. 1987, 1, 149–168. [Google Scholar]
- Masaki, T.; Igarashi, K.; Tokuda, M.; Yukimasa, S.; Han, F.; Jin, Y.J.; Li, J.Q.; Yoneyama, H.; Uchida, N.; Fujita, J.; et al. pp60c-src activation in lung adenocarcinoma. Eur. J. Cancer 2003, 39, 1447–1455. [Google Scholar] [CrossRef]
- Elsberger, B.; Fullerton, R.; Zino, S.; Jordan, F.; Mitchell, T.J.; Brunton, V.G.; Mallon, E.A.; Shiels, P.G.; Edwards, J. Breast cancer patients’ clinical outcome measures are associated with Src kinase family member expression. Br. J. Cancer 2010, 103, 899–909. [Google Scholar] [CrossRef]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem. 2009, 284, 36592–36604. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E. Src kinases are important regulators of mitochondrial functions. Int. J. Biochem. Cell Biol. 2013, 45, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.L.; Sun, H.F.; Gao, S.P.; Li, L.D.; Huang, S.; Hu, X.; Liu, S.; Wu, J.; Shao, Z.M.; Jin, W. SSBP1 Suppresses TGFbeta-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res. 2016, 76, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Montessuit, S.; Magnenat, E.; Vilbois, F.; Hochmann, A.; Journot, L.; Antonsson, B.; Martinou, J.C. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell 2001, 8, 601–611. [Google Scholar] [CrossRef]
- Izeradjene, K.; Douglas, L.; Delaney, A.B.; Houghton, J.A. Casein kinase I attenuates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by regulating the recruitment of fas-associated death domain and procaspase-8 to the death-inducing signaling complex. Cancer Res. 2004, 64, 8036–8044. [Google Scholar] [CrossRef]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, B.S.; Edzuma, A.N.; Hough, M.A.; Blundell, K.L.; Kagan, V.E.; Kapralov, A.A.; Fraser, L.A.; Butt, J.N.; Silkstone, G.G.; Wilson, M.T.; et al. The hydrogen-peroxide-induced radical behaviour in human cytochrome c-phospholipid complexes: Implications for the enhanced pro-apoptotic activity of the G41S mutant. Biochem. J. 2013, 456, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Diaz-Quintana, A.; Salzano, M.; Orzaez, M.; Perez-Paya, E.; Teixeira, M.; De la Rosa, M.A.; Diaz-Moreno, I. Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti-apoptotic switch. J. Biol. Inorg. Chem. 2011, 16, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Castellano, A.; Diaz-Quintana, A.; Perez-Mejias, G.; Elena-Real, C.A.; Gonzalez-Arzola, K.; Garcia-Maurino, S.M.; De la Rosa, M.A.; Diaz-Moreno, I. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 7955–7960. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb Perspect Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.O.; Agostino, M.; Dharmarajan, A.; Pervaiz, S. Cross Talk Between Cellular Redox State and the Antiapoptotic Protein Bcl-2. Antioxid Redox Signal. 2018, 29, 1215–1236. [Google Scholar] [CrossRef]
- Low, I.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef]
- Rayavarapu, R.R.; Heiden, B.; Pagani, N.; Shaw, M.M.; Shuff, S.; Zhang, S.; Schafer, Z.T. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J. Biol. Chem. 2015, 290, 8722–8733. [Google Scholar] [CrossRef]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-p110alpha inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef]
- Nalluri, S.; Ghoshal-Gupta, S.; Kutiyanawalla, A.; Gayatri, S.; Lee, B.R.; Jiwani, S.; Rojiani, A.M.; Rojiani, M.V. TIMP-1 Inhibits Apoptosis in Lung Adenocarcinoma Cells via Interaction with Bcl-2. PLoS ONE 2015, 10, e0137673. [Google Scholar] [CrossRef]
- Polzien, L.; Baljuls, A.; Rennefahrt, U.E.; Fischer, A.; Schmitz, W.; Zahedi, R.P.; Sickmann, A.; Metz, R.; Albert, S.; Benz, R.; et al. Identification of novel in vivo phosphorylation sites of the human proapoptotic protein BAD: Pore-forming activity of BAD is regulated by phosphorylation. J. Biol. Chem. 2009, 284, 28004–28020. [Google Scholar] [CrossRef] [PubMed]
- Bhakar, A.L.; Howell, J.L.; Paul, C.E.; Salehi, A.H.; Becker, E.B.; Said, F.; Bonni, A.; Barker, P.A. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J. Neurosci. 2003, 23, 11373–11381. [Google Scholar] [CrossRef] [PubMed]
- Donovan, N.; Becker, E.B.; Konishi, Y.; Bonni, A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 2002, 277, 40944–40949. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Minemoto, Y.; Zhang, J.; Liu, J.; Tang, F.; Bui, T.N.; Xiang, J.; Lin, A. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell 2004, 13, 329–340. [Google Scholar] [CrossRef]
- Moujalled, D.; Weston, R.; Anderton, H.; Ninnis, R.; Goel, P.; Coley, A.; Huang, D.C.; Wu, L.; Strasser, A.; Puthalakath, H. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep. 2011, 12, 77–83. [Google Scholar] [CrossRef]
- Putcha, G.V.; Le, S.; Frank, S.; Besirli, C.G.; Clark, K.; Chu, B.; Alix, S.; Youle, R.J.; LaMarche, A.; Maroney, A.C.; et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 2003, 38, 899–914. [Google Scholar] [CrossRef]
- Putcha, G.V.; Moulder, K.L.; Golden, J.P.; Bouillet, P.; Adams, J.A.; Strasser, A.; Johnson, E.M. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 2001, 29, 615–628. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Majewski, M.; Nieborowska-Skorska, M.; Salomoni, P.; Slupianek, A.; Reiss, K.; Trotta, R.; Calabretta, B.; Skorski, T. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 1999, 59, 2815–2819. [Google Scholar]
- Kennedy, D.; Mnich, K.; Oommen, D.; Chakravarthy, R.; Almeida-Souza, L.; Krols, M.; Saveljeva, S.; Doyle, K.; Gupta, S.; Timmerman, V.; et al. HSPB1 facilitates ERK-mediated phosphorylation and degradation of BIM to attenuate endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2017, 8, e3026. [Google Scholar] [CrossRef]
- Arokium, H.; Ouerfelli, H.; Velours, G.; Camougrand, N.; Vallette, F.M.; Manon, S. Substitutions of potentially phosphorylatable serine residues of Bax reveal how they may regulate its interaction with mitochondria. J. Biol. Chem. 2007, 282, 35104–35112. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Gao, F.; May, W.S.; Flagg, T.; Deng, X. Protein kinase Czeta abrogates the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2007, 282, 21268–21277. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L.; Ismail, F.; Azad, A.; Ternette, N.; Leverrier, S.; Edelmann, M.J.; Kessler, B.M.; Leigh, I.M.; Jackson, S.; Storey, A. Tyrosine dephosphorylation is required for Bak activation in apoptosis. EMBO J. 2010, 29, 3853–3868. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Fox, J.; Leverrier, S.; Storey, A. Blockade of the BAK hydrophobic groove by inhibitory phosphorylation regulates commitment to apoptosis. PLoS ONE 2012, 7, e49601. [Google Scholar] [CrossRef] [PubMed]
- Afreen, S.; Bohler, S.; Muller, A.; Demmerath, E.M.; Weiss, J.M.; Jutzi, J.S.; Schachtrup, K.; Kunze, M.; Erlacher, M. BCL-XL expression is essential for human erythropoiesis and engraftment of hematopoietic stem cells. Cell Death Dis. 2020, 11, 8. [Google Scholar] [CrossRef]
- Loo, L.S.W.; Soetedjo, A.A.P.; Lau, H.H.; Ng, N.H.J.; Ghosh, S.; Nguyen, L.; Krishnan, V.G.; Choi, H.; Roca, X.; Hoon, S.; et al. BCL-xL/BCL2L1 is a critical anti-apoptotic protein that promotes the survival of differentiating pancreatic cells from human pluripotent stem cells. Cell Death Dis. 2020, 11, 378. [Google Scholar] [CrossRef]
- Motoyama, N.; Wang, F.; Roth, K.A.; Sawa, H.; Nakayama, K.; Nakayama, K.; Negishi, I.; Senju, S.; Zhang, Q.; Fujii, S.; et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995, 267, 1506–1510. [Google Scholar] [CrossRef]
- Kharbanda, S.; Saxena, S.; Yoshida, K.; Pandey, P.; Kaneki, M.; Wang, Q.; Cheng, K.; Chen, Y.N.; Campbell, A.; Sudha, T.; et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J. Biol. Chem. 2000, 275, 322–327. [Google Scholar] [CrossRef]
- Wang, J.; Beauchemin, M.; Bertrand, R. Bcl-xL phosphorylation at Ser49 by polo kinase 3 during cell cycle progression and checkpoints. Cell Signal. 2011, 23, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Beauchemin, M.; Bertrand, R. Phospho-Bcl-x(L)(Ser62) plays a key role at DNA damage-induced G(2) checkpoint. Cell Cycle 2012, 11, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003, 538, 41–47. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; et al. Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Simizu, S.; Osada, H. The phosphorylation status and anti-apoptotic activity of Bcl-2 are regulated by ERK and protein phosphatase 2A on the mitochondria. FEBS Lett. 2004, 569, 249–255. [Google Scholar] [CrossRef]
- Breitschopf, K.; Haendeler, J.; Malchow, P.; Zeiher, A.M.; Dimmeler, S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: Molecular characterization of the involved signaling pathway. Mol. Cell Biol. 2000, 20, 1886–1896. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezawa, A.; Ichijo, H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar] [CrossRef]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef]
- Kobayashi, S.; Lee, S.H.; Meng, X.W.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Craig, R.W.; Kaufmann, S.H.; Gores, G.J. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J. Biol. Chem. 2007, 282, 18407–18417. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Hwang, C.S.; Yin, J.H.; Chen, S.D.; Yang, D.I. Oncostatin M-dependent Mcl-1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. Biochim. Biophys. Acta 2015, 1853, 2306–2325. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Ren, X.; Yang, L.; Lin, Y.; Wu, X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003, 115, 61–70. [Google Scholar] [CrossRef]
- Park, B. JNK1mediated phosphorylation of Smac/DIABLO at the serine 6 residue is functionally linked to its mitochondrial release during TNFalpha-induced apoptosis of HeLa cells. Mol. Med. Rep. 2014, 10, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.H.; Chun, K.S.; Kundu, J.; Park, B. Phosphorylation of Smac by Akt promotes the caspase-3 activation during etoposide-induced apoptosis in HeLa cells. Mol. Carcinog. 2015, 54, 83–92. [Google Scholar] [CrossRef]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef]
- Gamas, P.; Marchetti, S.; Puissant, A.; Grosso, S.; Jacquel, A.; Colosetti, P.; Pasquet, J.M.; Mahon, F.X.; Cassuto, J.P.; Auberger, P. Inhibition of imatinib-mediated apoptosis by the caspase-cleaved form of the tyrosine kinase Lyn in chronic myelogenous leukemia cells. Leukemia 2009, 23, 1500–1506. [Google Scholar] [CrossRef]
- Luciano, F.; Herrant, M.; Jacquel, A.; Ricci, J.E.; Auberger, P. The P54-cleaved form of the tyrosine kinase Lyn generated by caspases during BCR-induced cell death in B lymphoma acts as a negative regulator of apoptosis. Faseb J. 2003, 17, 711. [Google Scholar] [CrossRef]
- Contri, A.; Brunati, A.M.; Trentin, L.; Cabrelle, A.; Miorin, M.; Cesaro, L.; Pinna, L.A.; Zambello, R.; Semenzato, G.; Donella-Deana, A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J. Clin. Invest. 2005, 115, 369–378. [Google Scholar] [CrossRef]
- Mahon, F.X.; Hayette, S.; Lagarde, V.; Belloc, F.; Turcq, B.; Nicolini, F.; Belanger, C.; Manley, P.W.; Leroy, C.; Etienne, G.; et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008, 68, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Mochly-Rosen, D. The PKCdelta -Abl complex communicates ER stress to the mitochondria—An essential step in subsequent apoptosis. J. Cell Sci. 2008, 121, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotrasová, V.; Keresztesová, B.; Ondrovičová, G.; Bauer, J.A.; Havalová, H.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease. Life 2021, 11, 82. https://doi.org/10.3390/life11020082
Kotrasová V, Keresztesová B, Ondrovičová G, Bauer JA, Havalová H, Pevala V, Kutejová E, Kunová N. Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease. Life. 2021; 11(2):82. https://doi.org/10.3390/life11020082
Chicago/Turabian StyleKotrasová, Veronika, Barbora Keresztesová, Gabriela Ondrovičová, Jacob A. Bauer, Henrieta Havalová, Vladimír Pevala, Eva Kutejová, and Nina Kunová. 2021. "Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease" Life 11, no. 2: 82. https://doi.org/10.3390/life11020082
APA StyleKotrasová, V., Keresztesová, B., Ondrovičová, G., Bauer, J. A., Havalová, H., Pevala, V., Kutejová, E., & Kunová, N. (2021). Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease. Life, 11(2), 82. https://doi.org/10.3390/life11020082