
PINK1: A Bridge between Mitochondria and Parkinson’s Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondria Homeostasis
3. PINK1 Helps Maintain Mitochondrial Homeostasis
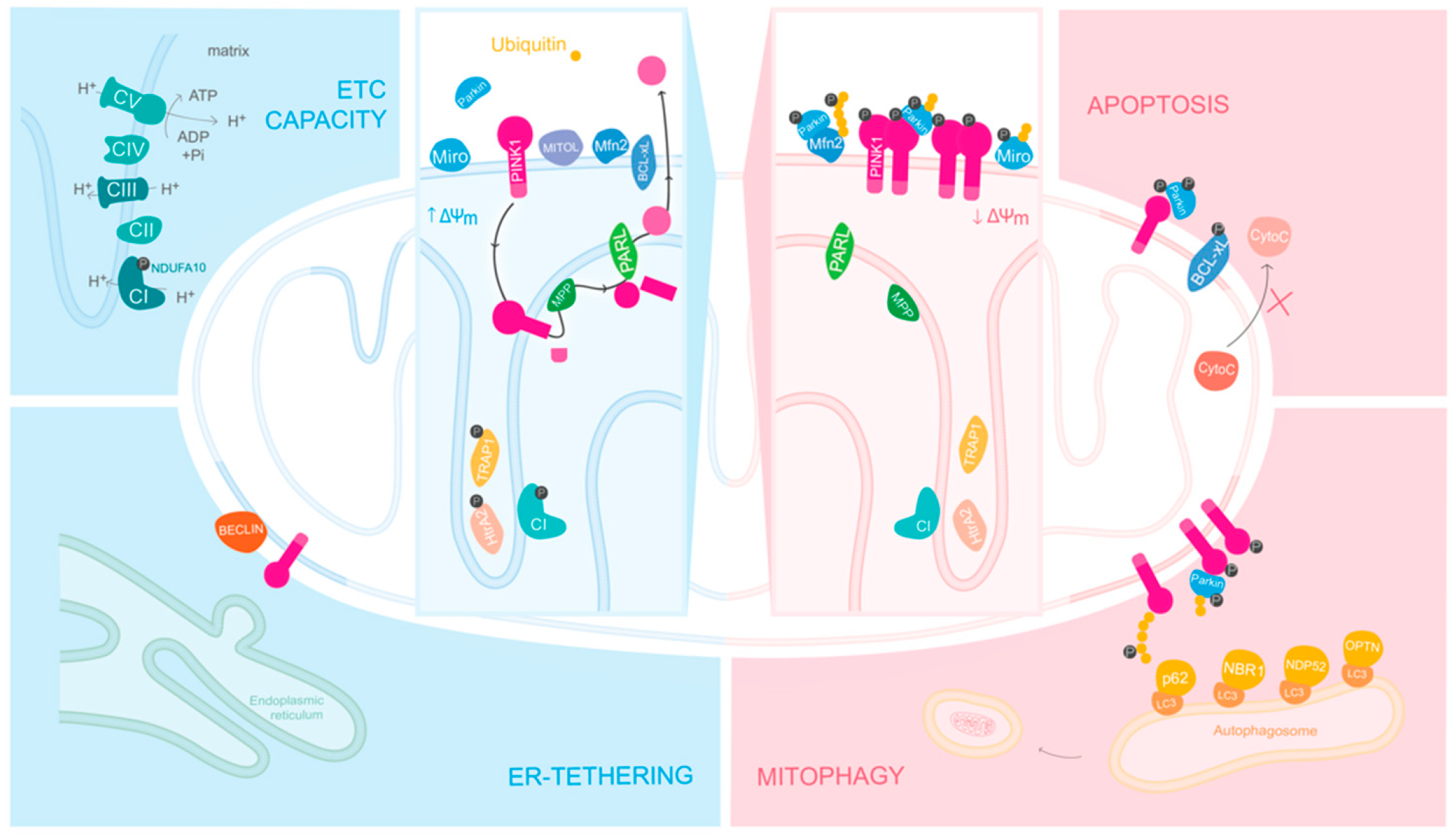
4. PINK1 in Healthy Mitochondria
5. PINK1 in Unhealthy Mitochondria
5.1. PINK1/Parkin Mediated Mitophagy
5.2. PINK1 Independent Mitophagy
6. PINK1 in PD Pathogenesis
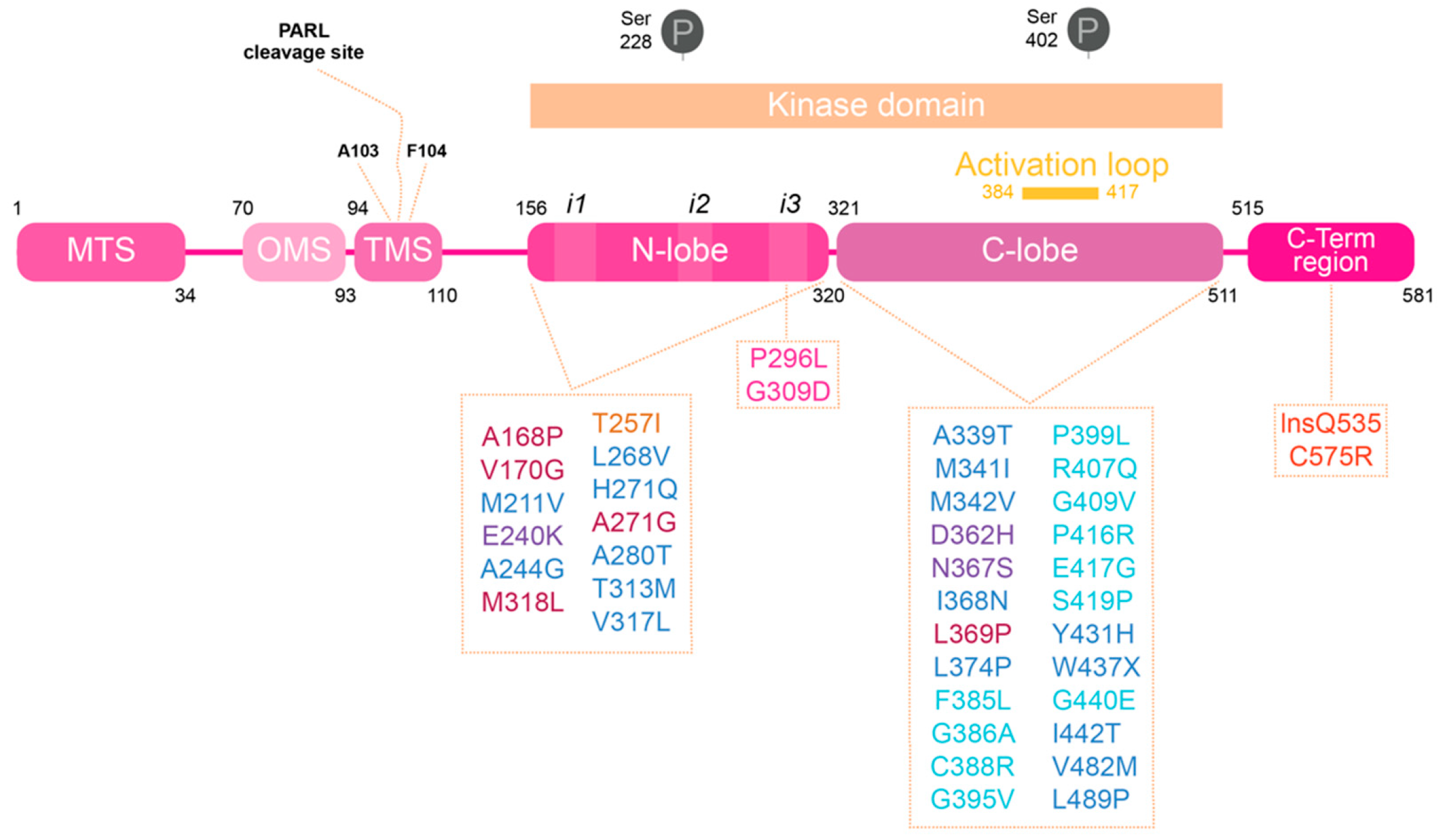
6.1. PINK1 as a Genetic Cause of PD
6.2. PINK1’s Link to Dysfunctions in the Respiratory Chain
6.3. PINK1 in Mitophagy Context
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig. 2013, 43, 886–893. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Am. J. Physiol.-Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kitagishi, Y.; Kobayashi, M. Function and Characteristics of PINK1 in Mitochondria. Oxidative Med. Cell. Longev. 2013, 2013, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef]
- Mei, Y.; Zhang, Y.; Yamamoto, K.; Xie, W.; Mak, T.W.; You, H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 5153–5158. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Tong, J.; Xu, Q.; Wu, Y.; Cai, F.; Li, T.; Song, W. Upregulation of human PINK1 gene expression by NFκB signalling. Mol. Brain 2014, 7, 57. [Google Scholar] [CrossRef]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Da Costa, C.A. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 2019, 73, 1028–1043.e5. [Google Scholar] [CrossRef]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef]
- Sim, C.H.; Lio, D.S.S.; Mok, S.S.; Masters, C.L.; Hill, A.F.; Culvenor, J.G.; Cheng, H.-C. C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet. 2006, 15, 3251–3262. [Google Scholar] [CrossRef]
- Beilina, A.; Van Der Brug, M.; Ahmad, R.; Kesavapany, S.; Miller, D.W.; Petsko, G.A.; Cookson, M.R. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 2005, 102, 5703–5708. [Google Scholar] [CrossRef] [PubMed]
- Woodroof, H.I.; Pogson, J.H.; Begley, M.; Cantley, L.C.; Deak, M.; Campbell, D.G.; van Aalten, D.M.F.; Whitworth, A.J.; Alessi, D.R.; Muqit, M.M.K. Discovery of catalytically active orthologues of the Parkinson’s disease kinase PINK1: Analysis of substrate specificity and impact of mutations. Open Biol. 2011, 1, 110012. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J. PINK 1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. In Vitro Comparison of the Activity Requirements and Substrate Specificity of Human and Triboleum castaneum PINK1 Orthologues. PLoS ONE 2016, 11, e0146083. [Google Scholar] [CrossRef]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. PINK1 Kinase Catalytic Activity Is Regulated by Phosphorylation on Serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef]
- Rasool, S.; Trempe, J.-F. New insights into the structure of PINK1 and the mechanism of ubiquitin phosphorylation. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 515–534. [Google Scholar] [CrossRef]
- Hua, G.; Zhang, Q.; Fan, Z. Heat Shock Protein 75 (TRAP1) Antagonizes Reactive Oxygen Species Generation and Protects Cells from Granzyme M-mediated Apoptosis. J. Biol. Chem. 2007, 282, 20553–20560. [Google Scholar] [CrossRef]
- Kadye, R.; Kramer, A.H.; Joos-Vandewalle, J.; Parsons, M.; Njengele, Z.; Hoppe, H.; Prinsloo, E. Guardian of the Furnace: Mitochondria, TRAP1, ROS and stem cell maintenance. IUBMB Life 2014, 66, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.-S.; Li, L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef] [PubMed]
- Plun-Favreau, H.; Klupsch, K.; Moisoi, N.; Gandhi, S.; Kjaer, S.; Frith, D.; Harvey, K.; Deas, E.; Harvey, R.J.; McDonald, N.; et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 2007, 9, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQuibban, A.; Vogel, H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 phosphorylates Drp1 S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H.V. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitofusins as mitochondrial anchors and tethers. J. Mol. Cell. Cardiol. 2020, 142, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Tsai, P.-I.; Course, M.M.; Lovas, J.R.; Hsieh, C.-H.; Babic, M.; Zinsmaier, K.E.; Wang, X. PINK1-mediated Phosphorylation of Miro Inhibits Synaptic Growth and Protects Dopaminergic Neurons in Drosophila. Sci. Rep. 2014, 4, 6962. [Google Scholar] [CrossRef]
- Williams, G.S.B.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Heeman, B.; Haute, C.V.D.; Aelvoet, S.-A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.H.; Willems, P.H.G.M.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Ludtmann, M.H.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1−/− zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Yi, M.; Weaver, D.; Hajnóczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2013, 106, 464–474. [Google Scholar] [CrossRef]
- Takatori, S.; Ito, G.; Iwatsubo, T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci. Lett. 2008, 430, 13–17. [Google Scholar] [CrossRef]
- Gandhi, S.; Muqit, M.M.K.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Richter, J.; Tocilescu, M.A.; Przedborski, S.; Voos, W. Pink1 Kinase and Its Membrane Potential (Δψ)-dependent Cleavage Product Both Localize to Outer Mitochondrial Membrane by Unique Targeting Mode. J. Biol. Chem. 2012, 287, 22969–22987. [Google Scholar] [CrossRef]
- Liu, Y.; Guardia-Laguarta, C.; Yin, J.; Erdjument-Bromage, H.; Martin, B.; James, M.; Jiang, X.; Przedborski, S. The Ubiquitination of PINK1 Is Restricted to Its Mature 52-kDa Form. Cell Rep. 2017, 20, 30–39. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Exner, N.; Treske, B.; Paquet, D.; Holmström, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.-H.; Gasser, T.; et al. Loss-of-Function of Human PINK1 Results in Mitochondrial Pathology and Can Be Rescued by Parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A Dimeric PINK1-containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef]
- Kitada, T.; Aakawa, S.; Hattori, N.; Matsumine, H.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosolmal recessive juvenile parkinsonism. Nat. Lett. 1998, 169, 166–169. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.-I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Trempe, J.-F.; Fon, E.A. Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front. Neurol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Komander, D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.P.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.-S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. P62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.T.; Wu, H.-C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 Ubiquitination of the Mitochondrial Transport Protein Miro Is Dependent on Serine 65 of the Parkin Ubiquitin Ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef] [PubMed]
- Safiulina, D.; Kuum, M.; Choubey, V.; Gogichaishvili, N.; Liiv, J.; Hickey, M.A.; Cagalinec, M.; Mandel, M.; Zeb, A.; Liiv, M.; et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019, 38, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Koyano, F.; Yamano, K.; Kosako, H.; Kimura, Y.; Kimura, M.; Fujiki, Y.; Tanaka, K.; Matsuda, N. Parkin-mediated ubiquitylation redistributes MITOL/March5 from mitochondria to peroxisomes. EMBO Rep. 2019, 20, e47728. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Martínez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar] [CrossRef]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of Phosphorylated ERK/MAP Kinases to Mitochondria and Autophagosomes in Lewy Body Diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J.; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 Function Promotes Mitophagy through Effects on Oxidative Stress and Mitochondrial Fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and Quality Control Defects in a Mouse Model of Gaucher Disease—Links to Parkinson’s Disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, srep44373. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- de Lau, L.M.L.; Breteler, M.M.B. The epidemiology of Parkinson’s disease. Lancet Neurol. 2006. [Google Scholar] [CrossRef]
- Albanese, A.; Valente, E.M.; Romito, L.M.; Bellacchio, E.; Elia, A.E.; Dallapiccola, B. The PINK1 phenotype can be indistinguishable from idiopathic Parkinson disease. Neurology 2005, 64, 1958–1960. [Google Scholar] [CrossRef]
- Koros, C.; Simitsi, A.; Stefanis, L. Genetics of Parkinson’s Disease: Genotype–Phenotype Correlations. Int. Rev. Neurobiol. 2017, 132, 197–231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tomiyama, H.; Sato, K.; Hatano, Y.; Yoshino, H.; Atsumi, M.; Kitaguchi, M.; Sasaki, S.; Kawaguchi, S.; Miyajima, H.; et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 2005, 64, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Hatano, Y.; Li, Y.; Sato, K.; Asakawa, S.; Yamamura, Y.; Tomiyama, H.; Bs, H.Y.; Asahina, M.; Kobayashi, S.; Hassin-Baer, S.; et al. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 2004, 56, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.F.; Gladkova, C.; Pardon, E.; Wagstaff, J.L.; Freund, S.M.V.; Steyaert, J.; Maslen, S.L.; Komander, D. Structure of PINK1 in complex with its substrate ubiquitin. Nature 2017, 552, 51–56. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.A.; Cartlidge, N.E.F.; Parker, W.D.; Turnbull, D.M. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Hoepken, H.-H.; Gispert, S.; Morales, B.; Wingerter, O.; Del Turco, D.; Mülsch, A.; Nussbaum, R.L.; Müller, K.; Dröse, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2007, 25, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Ripoli, M.; Quarato, G.; Scrima, R.; D’Aprile, A.; Boffoli, D.; Margaglione, M.; Criscuolo, C.; De Michele, G.; Sardanelli, A.; et al. Coexistence of mutations in PINK1 and mitochondrial DNA in early onset parkinsonism. J. Med. Genet. 2008, 45, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Sardanelli, A.; Scrima, R.; Ripoli, M.; Quarato, G.; D’Aprile, A.; Bellomo, F.; Scacco, S.; De Michele, G.; Filla, A.; et al. Mitochondrial Respiratory Dysfunction in Familiar Parkinsonism Associated with PINK1 Mutation. Neurochem. Res. 2008, 33, 2565–2574. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in Complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Rakovic, A.; Grünewald, A.; Seibler, P.; Ramirez, A.; Kock, N.; Orolicki, S.; Lohmann, K.; Klein, C. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum. Mol. Genet. 2010, 19, 3124–3137. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grünewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced Putative Kinase 1 (PINK1)-dependent ubiquitination of endogenous parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin Recruitment Is Impaired in Neurons Derived from Mutant PINK1 Induced Pluripotent Stem Cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee VM, Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Sliter, D.A.; Shammas, M.K.; Huang, X.; Wang, C.; Calvelli, H.; Maric, D.S.; Narendra, D.P. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 2021, 1–10. [Google Scholar] [CrossRef]
- Cornelissen, T.; Vilain, S.; Vints, K.; Gounko, N.; Verstreken, P.; Vandenberghe, W. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in drosophila. eLife 2018, 7, e35878. [Google Scholar] [CrossRef]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Benincá, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, F.B.; Morais, V.A. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life 2021, 11, 371. https://doi.org/10.3390/life11050371
Gonçalves FB, Morais VA. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life. 2021; 11(5):371. https://doi.org/10.3390/life11050371
Chicago/Turabian StyleGonçalves, Filipa Barroso, and Vanessa Alexandra Morais. 2021. "PINK1: A Bridge between Mitochondria and Parkinson’s Disease" Life 11, no. 5: 371. https://doi.org/10.3390/life11050371
APA StyleGonçalves, F. B., & Morais, V. A. (2021). PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life, 11(5), 371. https://doi.org/10.3390/life11050371