
The Role of Autophagy in Eye Diseases

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
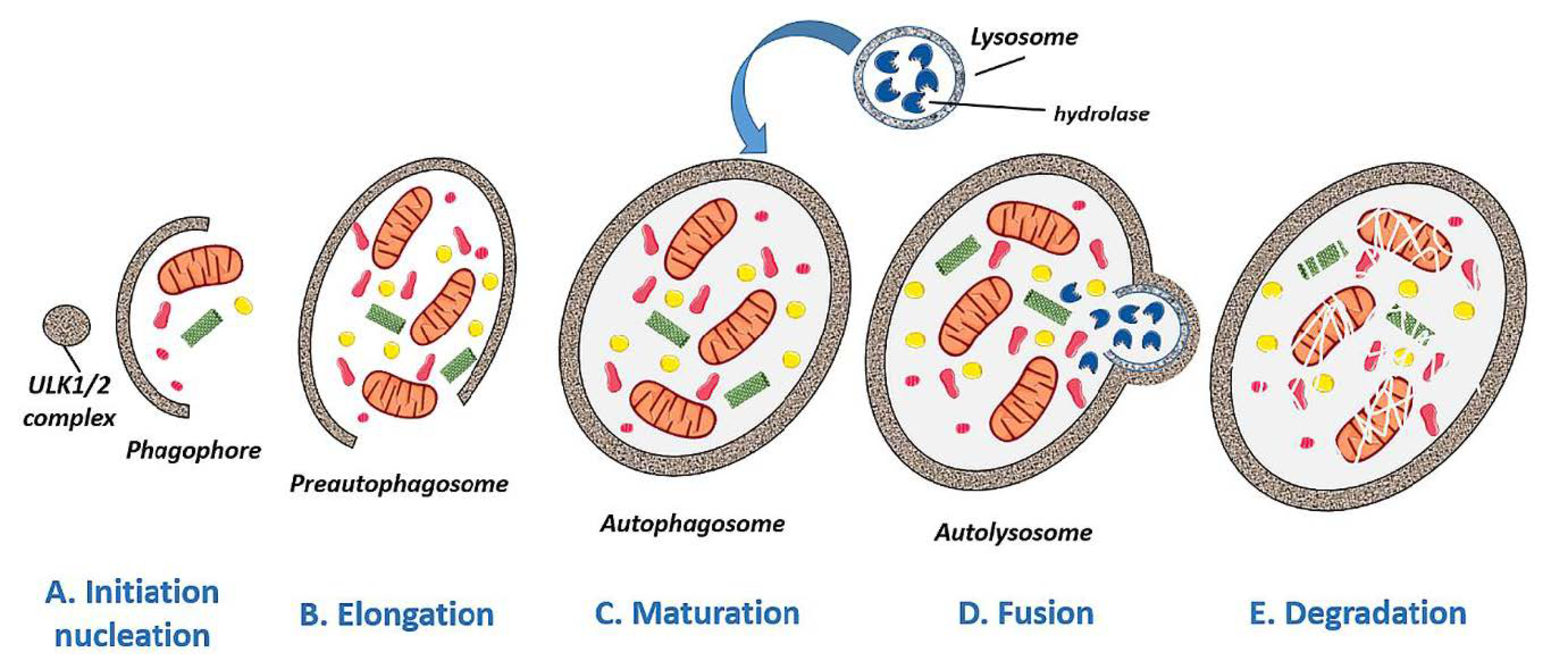
2. Overview of Autophagy
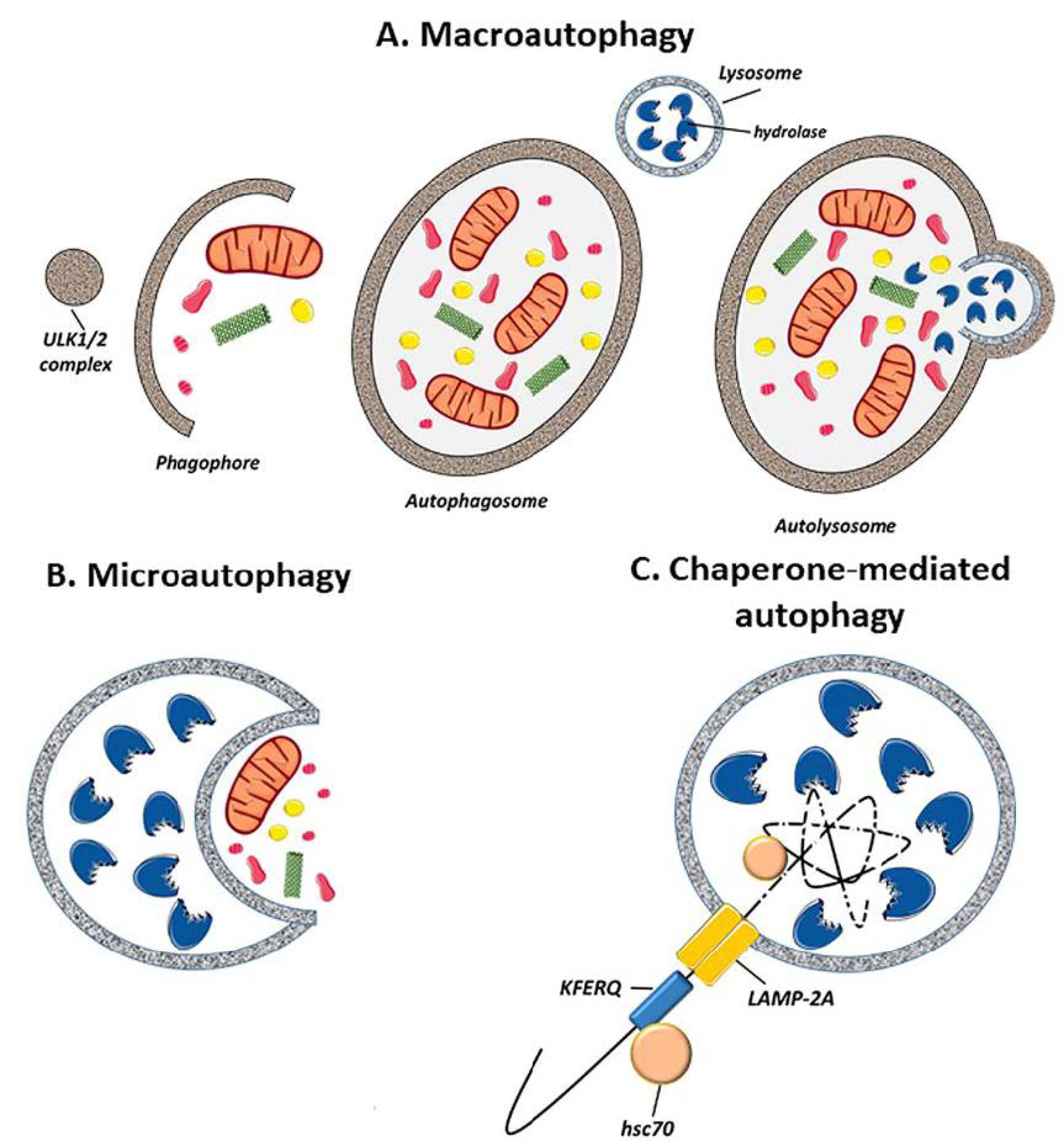
2.1. Autophagy Types
2.2. Regulation of Autophagy: Signaling Pathways
2.3. Physiological Functions of Autophagy
2.4. Pathological Implications of Autophagy
3. Autophagy in Ocular Diseases
3.1. Glaucoma
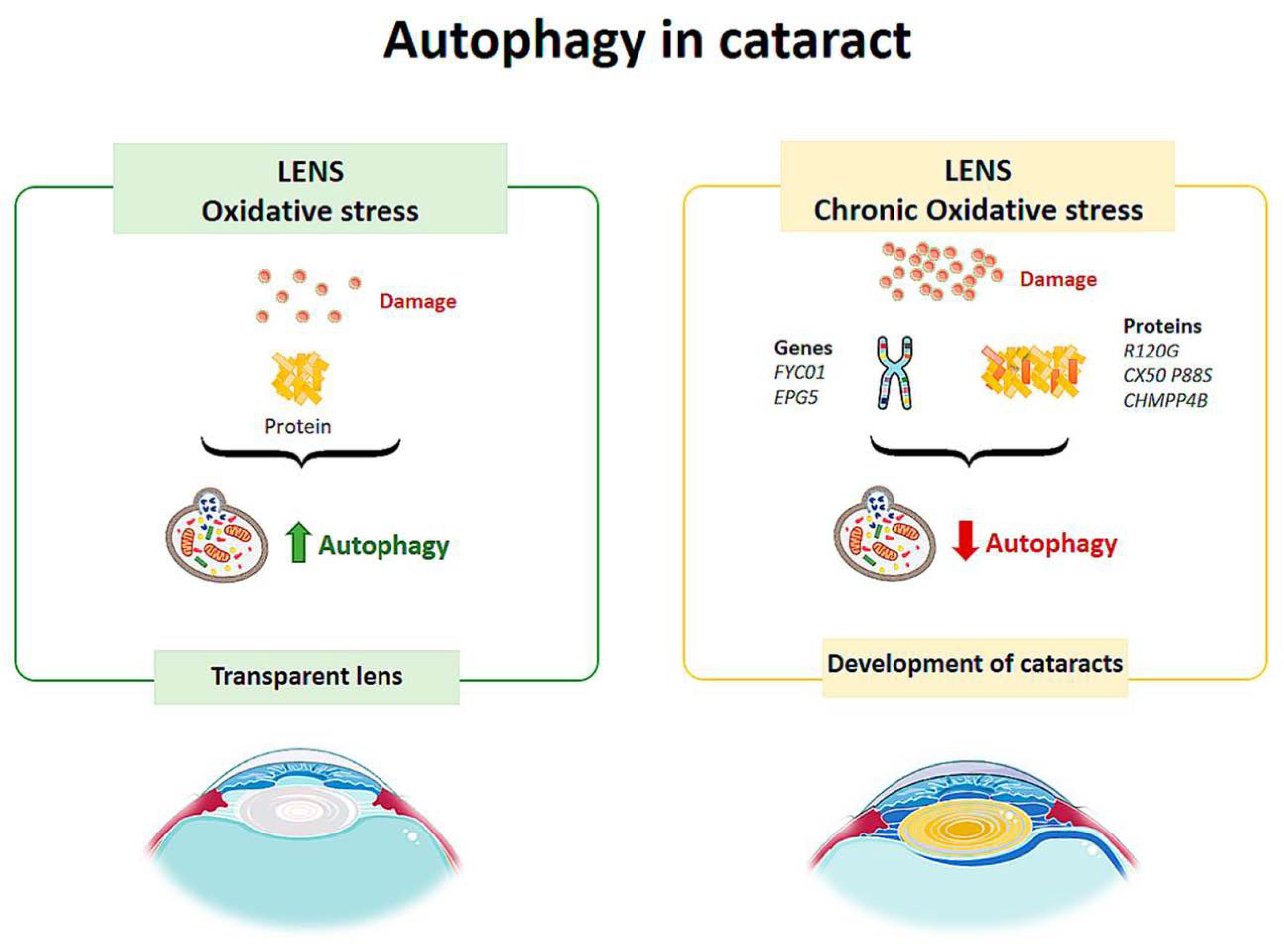
3.2. Cataract
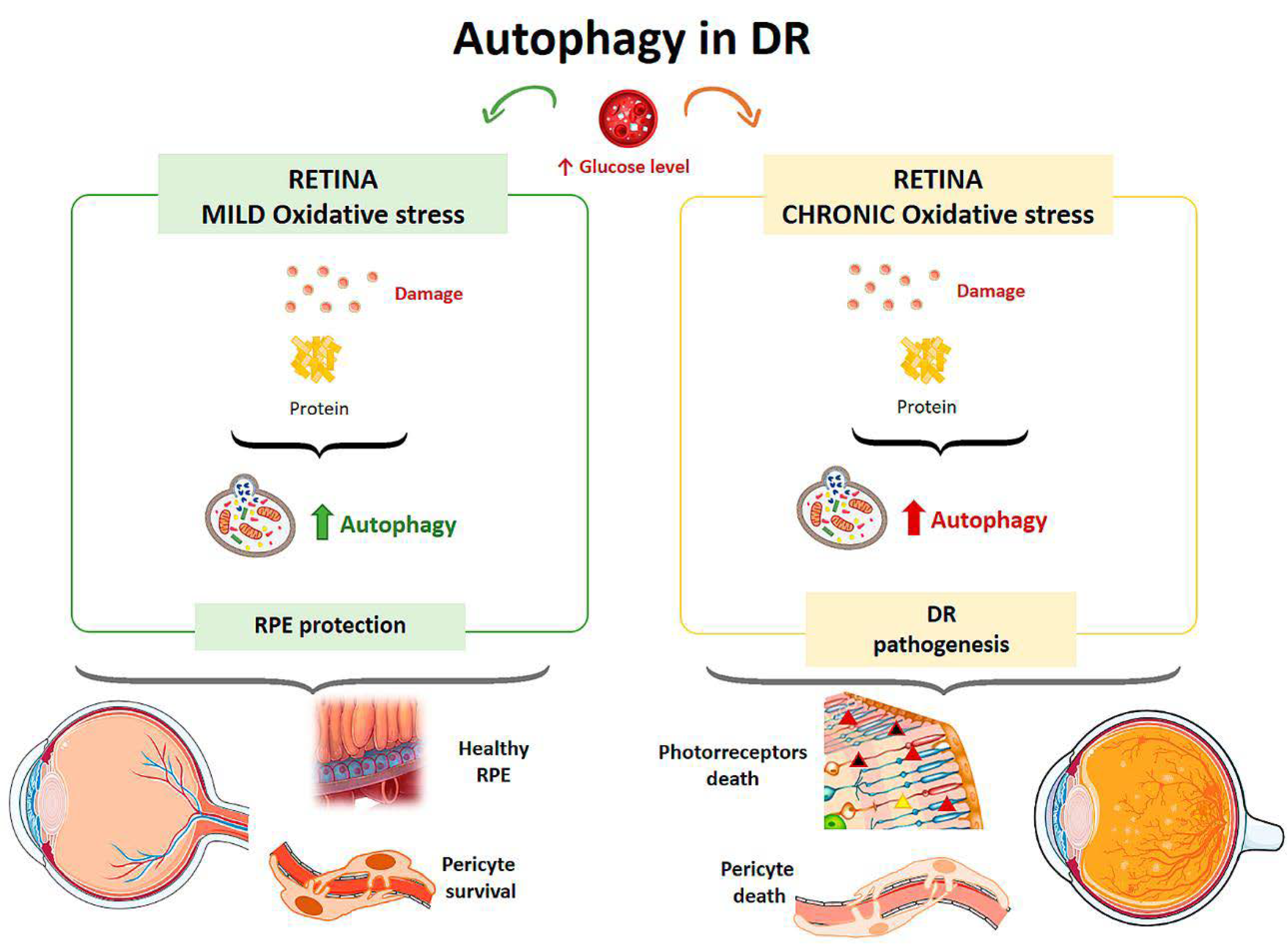
3.3. Diabetic Retinopathy
3.4. Age-Related Macular Degeneration
3.5. Autophagy as a Therapy in Ocular Pathologies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–312. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Viiri, J.; Amadio, M.; Marchesi, N.; Hyttinen, J.M.T.; Kivinen, N.; Sironen, R.; Rilla, K.; Akhtar, S.; Provenzani, A.; D’Agostino, V.G.; et al. Autophagy Activation Clears ELAVL1/HuR-Mediated Accumulation of SQSTM1/p62 during Proteasomal Inhibition in Human Retinal Pigment Epithelial Cells. PLoS ONE 2013, 8, e69563. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pan, X.; Zhao, X.; Luo, J.; Xu, M.; Bai, D.; Hu, Y.; Liu, X.; Yu, Q.; Gao, D. Autophagy and Age-Related Eye Diseases. Biomed. Res. Int. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Li, W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Cooley, P.L.; Dice, P.F. Corneal Dystrophy in the Dog and Cat. Vet. Clin. N. Am. Small Anim. Pract. 1990, 20, 681–692. [Google Scholar] [CrossRef]
- Kaden, T.R.; Li, W. Autophagy, Mitochondrial Dynamics, and Retinal Diseases. Asia-Pac. J. Ophthalmol. 2013, 2, 341–348. [Google Scholar] [CrossRef]
- Jin, M.; Liu, X.; Klionsky, D.J. SnapShot: Selective Autophagy. Cell 2013, 152, 368–368.e2. [Google Scholar] [CrossRef] [PubMed]
- Farré, J.-C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.-G. The Association of AMPK with ULK1 Regulates Autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two Distinct Vps34 Phosphatidylinositol 3–Kinase Complexes Function in Autophagy and Carboxypeptidase Y Sorting inSaccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Grimmel, M.; Backhaus, C.; Proikas-Cezanne, T. WIPI-Mediated Autophagy and Longevity. Cells 2015, 4, 202–217. [Google Scholar] [CrossRef]
- Zoukhri, D.; Fix, A.; Alroy, J.; Kublin, C.L. Mechanisms of Murine Lacrimal Gland Repair after Experimentally Induced Inflammation. Investig. Opthalmol. Vis. Sci. 2008, 49, 4399–4406. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein Turnover Via Autophagy: Implications for Metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Master, S.S.; De Haro, S.A.; Delgado, M.; Roberts, E.A.; Hope, J.C.; Keane, J.; Deretic, V. Th1–Th2 polarisation and autophagy in the control of intracellular mycobacteria by macrophages. Vet. Immunol. Immunopathol. 2009, 128, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, M.; Ozawa, Y.; Shinmura, K.; Inaba, T.; Nakamura, S.; Kawakita, T.; Watanabe, M.; Tsubota, K. Calorie restriction (CR) and CR mimetics for the prevention and treatment of age-related eye disorders. Exp. Gerontol. 2013, 48, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Kyoi, S.; Yamaguchi, O.; Sadoshima, J.; Otsu, K. The role of autophagy in the heart. Cell Death Differ. 2009, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Rehen, S.K.; Neves, D.D.C.; Fragel-Madeira, L.; Britto, L.R.G.; Linden, R. Selective sensitivity of early postmitotic retinal cells to apoptosis induced by inhibition of protein synthesis. Eur. J. Neurosci. 1999, 11, 4349–4356. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Veréb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Frost, L.S.; Mitchell, C.H.; Boesze-Battaglia, K. Autophagy in the eye: Implications for ocular cell health. Exp. Eye Res. 2014, 124, 56–66. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Villarejo-Zori, B.; Ball, G.; Boya, P.; Ganley, I.G. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy 2019, 15, 1296–1308. [Google Scholar] [CrossRef]
- Remé, C.; Wirz-Justice, A.; Rhyner, A.; Hofmann, S. Circadian rhythm in the light response of rat retinal disk-shedding and autophagy. Brain Res. 1986, 369, 356–360. [Google Scholar] [CrossRef]
- Yao, J.; Jia, L.; Shelby, S.J.; Ganios, A.M.; Feathers, K.; Thompson, D.A.; Zacks, D.N. Circadian and Noncircadian Modulation of Autophagy in Photoreceptors and Retinal Pigment Epithelium. Investig. Opthalmol. Vis. Sci. 2014, 55, 3237–3246. [Google Scholar] [CrossRef]
- Hornung, J.P.; Koppel, H.; Clarke, P.G.H. Endocytosis and autophagy in dying neurons: An ultrastructural study in chick embryos. J. Comp. Neurol. 1989, 283, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Karantza, V.; White, E. Role of Autophagy in Breast Cancer. Autophagy 2007, 3, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy 2009, 5, 563–564. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Munemasa, Y.; Kwong, J.M.K.; Ahn, J.H.; Mareninov, S.; Gordon, L.K.; Caprioli, J.; Piri, N. Activation of autophagy in retinal ganglion cells. J. Neurosci. Res. 2008, 86, 2943–2951. [Google Scholar] [CrossRef]
- Fu, C.T.; Sretavan, D. Involvement of EphB/ephrin-B signaling in axonal survival in mouse experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 76–84. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef]
- McMonnies, C.W. Hyperbaric oxygen therapy and the possibility of ocular complications or contraindications. Clin. Exp. Optom. 2015, 98, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Duran, M.D.; Shoaie-Nia, K.; Zanon-Moreno, V.; Sanz-Gonzalez, S.M.; del Castillo, J.B.; Garcia-Medina, J.J. Strategies to Reduce Oxidative Stress in Glaucoma Patients. Curr. Neuropharmacol. 2018, 16, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells. Autophagy 2013, 9, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Jeyabalan, N.; Liton, P.B. MTOR-independent induction of autophagy in trabecular meshwork cells subjected to biaxial stretch. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1054–1062. [Google Scholar] [CrossRef]
- Pulliero, A.; Seydel, A.; Camoirano, A.; Saccà, S.C.; Sandri, M.; Izzotti, A. Oxidative Damage and Autophagy in the Human Trabecular Meshwork as Related with Ageing. PLoS ONE 2014, 9, e98106. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Hirt, J.; Stamer, W.D.; Liton, P.B. Autophagic dysregulation in glaucomatous trabecular meshwork cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 379–385. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Davis, L.K.; Meyer, K.J.; Schindler, E.I.; Beck, J.S.; Rudd, D.S.; Grundstad, A.J.; Scheetz, T.E.; Braun, T.A.; Fingert, J.H.; Alward, W.L.M.; et al. Copy Number Variations and Primary Open-Angle Glaucoma. Investig. Opthalmol. Vis. Sci. 2011, 52, 7122–7133. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, M.S.; Fingert, J.H.; Roos, B.E.; Chen, S.; Holmes, R.; Graham, S.L.; Chehade, M.; Galanopolous, A.; Ridge, B.; Souzeau, E.; et al. Copy Number Variations of TBK1 in Australian Patients With Primary Open-Angle Glaucoma. Am. J. Ophthalmol. 2015, 159, 124–130.e1. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, K.; Kumari, A.; Radha, V.; Swarup, G. A Glaucoma-Associated Variant of Optineurin, M98K, Activates Tbk1 to Enhance Autophagosome Formation and Retinal Cell Death Dependent on Ser177 Phosphorylation of Optineurin. PLoS ONE 2015, 10, e0138289. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Rezaie, T. Adult-Onset Primary Open-Angle Glaucoma Caused by Mutations in Optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.-L.; Akahori, M.; Obazawa, M.; Minami, M.; Noda, T.; Nakaya, N.; Tomarev, S.; Kawase, K.; Yamamoto, T.; Noda, S.; et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum. Mol. Genet. 2010, 19, 2606–2615. [Google Scholar] [CrossRef]
- Chalasani, M.L.; Radha, V.; Gupta, V.; Agarwal, N.; Balasubramanian, D.; Swarup, G. A Glaucoma-Associated Mutant of Optineurin Selectively Induces Death of Retinal Ganglion Cells Which Is Inhibited by Antioxidants. Investig. Opthalmol. Vis. Sci. 2007, 48, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Boya, P. Why autophagy is good for retinal ganglion cells? Eye 2017, 31, 185–190. [Google Scholar] [CrossRef][Green Version]
- Rodríguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012, 19, 162–169. [Google Scholar] [CrossRef]
- Lin, W.; Kuang, H. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 2014, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Rosignol, I.; Sierra-Filardi, E.; Rodriguez-Muela, N.; Schmelter, C.; Cecconi, F.; Grus, F.; Boya, P. Age related retinal Ganglion cell susceptibility in context of autophagy deficiency. Cell Death Discov. 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.L.; Kim, J.H.; Park, C.K. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis. 2012, 3, e290–e299. [Google Scholar] [CrossRef] [PubMed]
- Knoferle, J.; Koch, J.C.; Ostendorf, T.; Michel, U.; Planchamp, V.; Vutova, P.; Tonges, L.; Stadelmann, C.; Bruck, W.; Bahr, M.; et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 6064–6069. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Kang, Q.; Ma, B.; Gao, S.; Li, X.; Liu, Y. Activation of autophagy and paraptosis in retinal ganglion cells after retinal ischemia and reperfusion injury in rats. Exp. Ther. Med. 2015, 9, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.; Gianetto, D.; Conte, D.; Bosone, A.; Vercelli, A. Activation of Autophagy in a Rat Model of Retinal Ischemia following High Intraocular Pressure. PLoS ONE 2011, 6, e22514. [Google Scholar] [CrossRef]
- Russo, R.; Berliocchi, L.; Adornetto, A.; Varano, G.P.; Cavaliere, F.; Nucci, C.; Rotiroti, D.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011, 2, e144. [Google Scholar] [CrossRef]
- Russo, R.; Varano, G.P.; Adornetto, A.; Nazio, F.; Tettamanti, G.; Girardello, R.; Cianfanelli, V.; Cavaliere, F.; Morrone, L.A.; Corasaniti, M.T.; et al. Rapamycin and fasting sustain autophagy response activated by ischemia/reperfusion injury and promote retinal ganglion cell survival. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Nucci, C.; Corasaniti, M.T.; Bagetta, G.; Morrone, L.A. Autophagy dysregulation and the fate of retinal ganglion cells in glaucomatous optic neuropathy. Prog. Brain Res. 2015, 220, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.J.; Brennan, L.A.; Basu, S.; Chauss, D.; Mohamed, A.; Gilliland, K.O.; Johnsen, S.; Menko, A.S.; Kantorow, M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013, 116, 141–150. [Google Scholar] [CrossRef]
- Zhou, J.; Yao, K.; Zhang, Y.; Chen, G.; Lai, K.; Yin, H.; Yu, Y. Thioredoxin Binding Protein-2 Regulates Autophagy of Human Lens Epithelial Cells under Oxidative Stress via Inhibition of Akt Phosphorylation. Oxid. Med. Cell. Longev. 2016, 2016, 1–17. [Google Scholar] [CrossRef]
- Sidjanin, D.J.; Park, A.K.; Ronchetti, A.; Martins, J.; Jackson, W.T. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016, 12, 1759–1775. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Eguchi, S.; Kimura, H.; Sasaki, J.; Sakamaki, Y.; Robinson, M.L.; Sasaki, T.; Mizushima, N. Deletion of Autophagy-related 5 (Atg5) and Pik3c3 Genes in the Lens Causes Cataract Independent of Programmed Organelle Degradation. J. Biol. Chem. 2013, 288, 11436–11447. [Google Scholar] [CrossRef]
- Chen, J.; Ma, Z.; Jiao, X.; Fariss, R.; Kantorow, W.L.; Kantorow, M.; Pras, E.; Frydman, M.; Pras, E.; Riazuddin, S.; et al. Mutations in FYCO1 Cause Autosomal-Recessive Congenital Cataracts. Am. J. Hum. Genet. 2011, 88, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Wignes, J.A.; Goldman, J.W.; Weihl, C.C.; Bartley, M.G.; Andley, U.P. p62 expression and autophagy in αB-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp. Eye Res. 2013, 115, 263–273. [Google Scholar] [CrossRef]
- Ge, X.-L.; Zhang, Y.; Wu, Y.; LV, J.; Zhang, W.; Jin, Z.-B.; Qu, J.; Gu, F. Identification of a Novel GJA8 (Cx50) Point Mutation Causes Human Dominant Congenital Cataracts. Sci. Rep. 2015, 4, 4121. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Sagona, A.P.; Nezis, I.P.; Stenmark, H. Association of CHMP4B and Autophagy with Micronuclei: Implications for Cataract Formation. Biomed. Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Volpe, C.M.O.; Villar-Delfino, P.H.; dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Fanjul-Moles, M.L.; López-Riquelme, G.O. Relationship between Oxidative Stress, Circadian Rhythms, and AMD. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Piano, I.; Novelli, E.; Della Santina, L.; Strettoi, E.; Cervetto, L.; Gargini, C. Involvement of autophagic pathway in the progression of retinal degeneration in a mouse model of diabetes. Front. Cell. Neurosci. 2016, 10, 1–12. [Google Scholar] [CrossRef]
- Yao, J.; Tao, Z.F.; Li, C.P.; Li, X.M.; Cao, G.F.; Jiang, Q.; Yan, B. Regulation of autophagy by high glucose in human retinal pigment epithelium. Cell. Physiol. Biochem. 2014, 33, 107–116. [Google Scholar] [CrossRef]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tikhonenko, M.; Bozack, S.N.; Lydic, T.A.; Yan, L.; Panchy, N.L.; Mcsorley, K.M.; Faber, M.S.; Yan, Y.; Boulton, M.E.; et al. Changes in the Daily Rhythm of Lipid Metabolism in the Diabetic Retina. PLoS ONE 2014, 9, e95028. [Google Scholar] [CrossRef]
- Wang, Q.; Bozack, S.N.; Yan, Y.; Boulton, M.E.; Grant, M.B.; Busik, J.V. Regulation of Retinal Inflammation by Rhythmic Expression of MiR-146a in Diabetic Retina. Investig. Opthalmol. Vis. Sci. 2014, 55, 3986–3994. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, I.A.D.; Lewin, I.G.; O’Hare, J.P.; Arendt, J.; Corrall, R.J.M. Abnormal circadian rhythm of melatonin i ndiabetic autonomic neuropathy. Clin. Endocrinol. 1986, 24, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Bao, X.L.; Cong, Y.Y.; Fan, B.; Li, G.Y.; Žerovnik, E. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Godley, B.F.; Shamsi, F.A.; Liang, F.-Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue Light Induces Mitochondrial DNA Damage and Free Radical Production in Epithelial Cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.-Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA Damage and Repair in RPE Associated with Aging and Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 3521–3529. [Google Scholar] [CrossRef]
- Szatmári-Tóth, M.; Kristóf, E.; Veréb, Z.; Akhtar, S.; Facskó, A.; Fésüs, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef]
- Szatmári-Tóth, M.; Ilmarinen, T.; Mikhailova, A.; Skottman, H.; Kauppinen, A.; Kaarniranta, K.; Kristóf, E.; Lytvynchuk, L.; Veréb, Z.; Fésüs, L.; et al. Human embryonic stem cell-derived retinal pigment epithelium-role in dead cell clearance and inflammation. Int. J. Mol. Sci. 2019, 20, 926. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ji, L.; Li, L.; Li, J. Oxidative stress, autophagy and pyroptosis in the neovascularization of oxygen‑induced retinopathy in mice. Mol. Med. Rep. 2018, 19, 927–934. [Google Scholar] [CrossRef]
- Holz, F.; Schütt, F.; Kopitz, J.; Eldred, G.; Kruse, F.; Völcker, H.; Cantz, M. Inhibition of Lysosomal Degradative Functions in RPE Cells by a Retinoid Component of Lipofuscin. Investig. Ophthalmol. Vis. Sci. 1999, 40, 737–743. [Google Scholar] [PubMed]
- De Jong, P.T.V.M. Drusen, AMD, and history. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 2061–2062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J.; Bai, Y.; Huang, L.; Qi, Y.; Zhang, Q.; Li, S.; Wu, Y.; Li, X. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: Implications for age-related macular degeneration. Cell Death Dis. 2015, 6, e1972. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Salminen, A.; Eskelinen, E.L.; Kopitz, J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2009, 8, 128–139. [Google Scholar] [CrossRef]
- Boya, P.; González-Polo, R.-A.; Casares, N.; Perfettini, J.-L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of Macroautophagy Triggers Apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and Exosomes in the Aged Retinal Pigment Epithelium: Possible Relevance to Drusen Formation and Age-Related Macular Degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Piippo, N.; Hytti, M.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Prevention of autophagy activates inflammasome signaling in ARPE-19 cells treated with a proteasome inhibitor. Acta Ophthalmol. 2013, 91. [Google Scholar] [CrossRef]
- Liu, J.; Copland, D.A.; Theodoropoulou, S.; Chiu, H.A.A.; Barba, M.D.; Mak, K.W.; Mack, M.; Nicholson, L.B.; Dick, A.D. Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep. 2016, 6, 20639. [Google Scholar] [CrossRef]
- Wang, L.; Cano, M.; Handa, J.T. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1248–1258. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.-L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef]
- Valapala, M.; Edwards, M.; Hose, S.; Grebe, R.; Bhutto, I.A.; Cano, M.; Berger, T.; Mak, T.W.; Wawrousek, E.; Handa, J.T.; et al. Increased Lipocalin-2 in the retinal pigment epithelium of Cryba1 cKO mice is associated with a chronic inflammatory response. Aging Cell 2014, 13, 1091–1094. [Google Scholar] [CrossRef]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef]
- Sinha, D.; Klise, A.; Sergeev, Y.; Hose, S.; Bhutto, I.A.; Hackler, L.; Malpic-llanos, T.; Samtani, S.; Grebe, R.; Goldberg, M.F.; et al. βA3/A1-crystallin in astroglial cells regulates retinal vascular remodeling during development. Mol. Cell. Neurosci. 2008, 37, 85–95. [Google Scholar] [CrossRef]
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; de la Villa, P.; de la Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 2013, 12, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, Q.; Cao, G.; Yao, J.; Yan, B. Repertoires of Autophagy in the Pathogenesis of Ocular Diseases. Cell. Physiol. Biochem. 2015, 35, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin Is Neuroprotective in a Rat Chronic Hypertensive Glaucoma Model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Petrovski, G.; Salminen, A.; Kaarniranta, K. 5′-Adenosine Monophosphate-Activated Protein Kinase–Mammalian Target of Rapamycin Axis As Therapeutic Target for Age-Related Macular Degeneration. Rejuvenation Res. 2011, 14, 651–660. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and Autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef]
- Fu, J.; Shao, C.-J.; Chen, F.-R.; Ng, H.-K.; Chen, Z.-P. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro. Oncol. 2010, 12, 328–340. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, H.; Zhang, Y.; Zhou, Z.; Wu, S. MicroRNA-29 enhances autophagy and cleanses exogenous mutant αB-crystallin in retinal pigment epithelial cells. Exp. Cell Res. 2019, 374, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Liang, X.; Peng, Y.; Yu, D.; Zhang, R.; Jin, H.; Fan, J.; Cai, W.; Ren, C.; Yu, J. 17β-estradiol ameliorates oxidative stress and blue light-emitting diode-induced retinal degeneration by decreasing apoptosis and enhancing autophagy. Drug Des. Devel. Ther. 2018, 12, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Huang, T.Y.; Chen, H.Y.; Huang, T.C.; Lin, L.C.; Chang, Y.J.; Hsia, S.M. Protective Effect of Melatonin against Oxidative Stress-Induced Apoptosis and Enhanced Autophagy in Human Retinal Pigment Epithelium Cells. Oxid. Med. Cell. Longev. 2018, 2018, 9015765. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, E.; Szczepanska, J.; Koskela, A.; Kaarniranta, K.; Blasiak, J. Dietary polyphenols in age-related macular degeneration: Protection against oxidative stress and beyond. Oxid. Med. Cell. Longev. 2019, 2019, 9682318. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, X.; Liu, L.; Shu, W.; Huang, Z. Sodium tanshinone IIA sulfonate protects ARPE-19 cells against oxidative stress by inhibiting autophagy and apoptosis. Sci. Rep. 2018, 8, 15137. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Albarral, J.A.; de Julián-López, E.; Soler-Domínguez, C.; de Hoz, R.; López-Cuenca, I.; Salobrar-García, E.; Ramírez, J.M.; Pinazo-Durán, M.D.; Salazar, J.J.; Ramírez, A.I. The Role of Autophagy in Eye Diseases. Life 2021, 11, 189. https://doi.org/10.3390/life11030189
Fernández-Albarral JA, de Julián-López E, Soler-Domínguez C, de Hoz R, López-Cuenca I, Salobrar-García E, Ramírez JM, Pinazo-Durán MD, Salazar JJ, Ramírez AI. The Role of Autophagy in Eye Diseases. Life. 2021; 11(3):189. https://doi.org/10.3390/life11030189
Chicago/Turabian StyleFernández-Albarral, José A., Esther de Julián-López, Carmen Soler-Domínguez, Rosa de Hoz, Inés López-Cuenca, Elena Salobrar-García, José M. Ramírez, María D. Pinazo-Durán, Juan J. Salazar, and Ana I. Ramírez. 2021. "The Role of Autophagy in Eye Diseases" Life 11, no. 3: 189. https://doi.org/10.3390/life11030189
APA StyleFernández-Albarral, J. A., de Julián-López, E., Soler-Domínguez, C., de Hoz, R., López-Cuenca, I., Salobrar-García, E., Ramírez, J. M., Pinazo-Durán, M. D., Salazar, J. J., & Ramírez, A. I. (2021). The Role of Autophagy in Eye Diseases. Life, 11(3), 189. https://doi.org/10.3390/life11030189