
Complete Mitochondrial Genome of Trichuristrichiura from Macaca sylvanus and Papio papio



Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Seville, Calle San Fernando, 4, 41004 Sevilla, Spain
*
Author to whom correspondence should be addressed.
Life 2021, 11(2), 126; https://doi.org/10.3390/life11020126
Submission received: 9 January 2021
/
Revised: 29 January 2021
/
Accepted: 3 February 2021
/
Published: 6 February 2021
(This article belongs to the Special Issue Trichuris: Other World, Other Life)
Abstract
:Trichuriasis is among the most prevalent worldwide parasitism caused by helminths. For many years, Trichuris spp. have been described with a relatively narrow range of both morphological and biometrical features. The use of the complete mitochondrial genome (mitogenome) is an alternative and powerful molecular method for inferring phylogenies. Here, we present an overview of the contributions of mitogenome for Trichuris spp. from human and non-human primates. In addition, we carry out structural and phylogenetic comparative analyses with genomes of Trichuris species available in public datasets. The complete mt genomes of Trichuris trichiura and Trichuris sp. from Macaca sylvanus and T. trichiura from Papio papio are 14,091 bp, 14,047 bp and 14,089 bp in length, respectively. The three mt genomes are circular and consist of 37 genes—13 PCGs (cox1–3, nad1–6, nad4L, atp6, atp8 and cob), 22 transfer RNA genes (tRNAs), and two rRNAs (rrnL and rrnS). The molecular evidence presented here supports the hypothesis that T. trichiura de M. sylvanus (TMF31) and T. trichiura de P. papio (TPM1) were similar but genetically different with respect to Trichuris sp. from macaques (TMM5). The phylogenetic study also supported the evolution of the different Trichuris species. In conclusion, we suggest the existence of two cryptic species parasitizing M. sylvanus.
1. Introduction
Soil-transmitted helminths (STHs) (Ascaris lumbricoides (Ascariasis), Trichuris trichiura (Trichuriasis)) and hookworms (Ancylostomiasis/Necatoriasis) are widely distributed in tropical and subtropical areas, with the greatest numbers occurring in sub-Saharan Africa, the Americas, China, and East Asia [1]. In areas with favorable climatic and environmental conditions, poor access to potable water supply, sanitation, and hygiene resources, the transmission of these parasites is higher [2]. Recent data show that about 1.5 billion people are estimated to be affected by STHs worldwide [1].
Although Trichuriasis is a seriously neglected disease, fortunately, in recent years hereditary studies are increasing as well as related methodologies that provide new opportunities for the discovery of novel intervention strategies, with major implications for improving animal and human health and welfare globally [3]. In addition, the implications of genomic studies could also be very relevant in relation to the search for new treatments for immunopathological diseases in humans [4,5,6,7,8,9]. For example, it has been reported that infections of human patients suffering from immunological disorders (such as Crohn’s disease) suppress clinical symptoms significantly with pig-Trichuris eggs [6,10,11,12].
The nematode mitogenome (complete mitochondrial genome) has several practical strengths as a phylogenetic marker, and has yielded well-supported results for clades, which were not well resolved using other approaches [13]. The whole genome and transcriptome studies have contributed to our understanding of deep node nematode phylogeny but have been limited by low taxon sampling (which is also biased toward some parasitic groups) and by the technical challenges inherent in obtaining a large quantity of genetic information from a single nematode with a small body size. In such cases, the use of mitogenome sequences is one alternative that has been widely applied to many nematode branches where relationships were unclear [13].
Nematode mitogenomes are like those of other animals in many respects, but have a few unusual features including high variation in conservation of gene order across major branches and the occasional presence of multiple chromosomes [13]. The nematode mitogenome is usually a single, circular molecule ranging in size from 12 to 22 kb and containing 36 (sometimes 37) genes—12 (or 13) protein coding genes (PCGs) (cox1–cox3, cytb, nad1-nad6, nad4L, atp6, and rarely atp8), two ribosomal RNA (rRNA) (rrnL and rrnS), and 22 transfer RNA genes (tRNAs). The atp8 gene, which is found in most other metazoan mitogenomes (except the parasitic Platyhelminthes clade Neodermata) [14], is usually absent in nematode mitogenomes, although it does appear in the order Trichinellida (Trichinella spp. and Trichuris spp.) [13,15,16,17,18,19,20]. Furthermore, within class Enoplea, members of order Trichinellida (Trichinella spp., Trichuris spp.) show a substantial gene rearrangement even among closely related species, while members of Chromadorea show far less rearrangement in theirs mitogenomes [21].
Kern et al. [13] concluded that the mitochondrial genome is a useful tool for nematode phylogenetic because the diversity within nematode mitogenome architecture, its variable rate of gene rearrangement, and the representation of nearly every kind of lifestyle and habitat ecology within nematodes make this phylum an exciting area for addressing questions about mitogenome evolution.
Currently, the complete mt genome of several species of whipworms has been sequenced—Trichuris trichiura from humans and baboons [3,19]; Trichuris sp. from the Endangered François’ Leaf-Monkey [17]; Trichuris rhinopiptheroxella from the endangered golden snub-nosed monkey [20]; Trichuris suis from pig [3,19]; Trichuris ovis from antelope [16]; Trichuris discolor from wild yak [16]; Trichuris skrjabini from sheep [22]; and Trichuris muris (LC050561, unpublished). This fact, and considering the hypothesis that Trichuris infecting primates represent a complex of cryptic species with some species capable of infecting both humans and non-human primates (NHPs), reflects the need to delve into the mt genomes of Trichuris spp. of NHPs compared to T. trichiura from humans.
Thus, the aim of the present study was to sequence the complete mt genome of Trichuris trichiura isolated from different NHPs. In addition, we carry out structural and phylogenetic comparative analyses with genomes of species of genus Trichuris available in public datasets.
2. Materials and Methods
2.1. Ethics Statement
This study does not require approval by an ethics committee. Macaca sylvanus, from which Trichuris specimens were collected from their caeca post-mortem. Trichuris specimens, recovered from the feces of one Papio papio after anthelmintic treatment, were handled and housed in a zoo in strict accordance with good animal practices.
2.2. Parasites, DNA Extraction and Genotyping of Worms
Specimen Trichuris worms were recollected from a Guinea baboon (P. papio) at Parque de la Naturaleza de Cabárceno (Cantabria, Spain), from a stool sample after anthelmintic treatment, and a Barbary macaque (M. sylvanus) in Castellar Zoo (Cádiz, Spain), from its caeca post-mortem. Adult Trichuris were recovered and washed in physiological saline, identified morphologically, and then, stored at −20 °C until use. Total genomic DNA was isolated from three individual worms according to the manufacturer’s protocol the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Düsseldorf, Germany),used to extract the genomic DNA. Quality of extractions was assessed using 0.8% agarose gel electrophoresis infused with SYBR® Safe DNA gel stain (Thermo Fisher Scientific, Waltham, MA, USA)
2.3. Mitochondrial Genome Amplification and Sequencing
For long-range PCR amplification and next generation sequencing (NGS), different primate derived Trichuris specimens were chosen. One Guinea baboon worm (TPM1) and two Barbary macaque worms (TMF31 and TMM5) were chosen based on their distinct haplotypes identified in previous studies by sequencing the partial gene of mtDNA (cox1, cob and rrnL) and rDNA (ITS1 and ITS2) [23]. To obtain the complete mt genome, firstly, we used the primers designed by Hawash et al. [19] to amplify the Trichuris sp. from baboon (TTB1) and T. trichiura from humans from Uganda (TTHUG) genomes in two overlapping fragments (~8 and ~6 kbp), Nevertheless, we only could amplify the second fragment (~6 kbp), from rrnL to nad1. Then, new primers (MS1F and MS1R) were designed to amplify the other fragment using Primer3 from nad1 to rrnL (http://bioinfo.ut.ee/primer3-0.4.0/ (accessed on 1 February 2021)). PCR mix, PCR conditions and PCR primers are summarized in the Supplementary materials (Table S1). The PCR products were checked on SYBR® Safe stained 0.8% Tris-Borate-EDTA (TBE) agarose gels and bands were eluted and purified from the agarose gel using the QWizard SV Gel and PCR Clean Up System Kit (Promega, Madison, WI, USA). Once purified, the samples were concentrated and measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Stab Vida (Lisbon, Portugal) sequenced the mt genomes. The PCR products were used for library construction using Illumina Nextera XT library preparation kit (San Diego, CA, USA) and the generated DNA fragments were sequenced in the Illumina MiSeq platform (San Diego, CA, USA), using 300 bp paired-end sequencing reads.
2.4. Assembly, Annotation, and Genome Sequence Analyses
Sequences were assembled and analyzed using MacVector package v17.5.4 (Oxford Molecular Group, Waterbeach, Cambridge, UK). The identity of the sequences was made using BLAST by comparison with other sequences available in GenBank database. Genome annotation was performed using the pipeline MITOS web server (http://mitos.bioinf.uni-leipzig.de/index.py (accessed on 1 February 2021)) [24] using the mitochondrial invertebrate genetic code, and MacVector and protein-encoding genes of T. trichiura. The majority of the tRNA genes were identified using the ARWEN tool (http://130.235.244.92/ARWEN/ (accessed on 1 February 2021)) to get rRNA [25] and tRNAScan-SE web server (http://trna.ucsc.edu/tRNAscan-SE/ (accessed on 1 February 2021)) [26], but the remaining tRNA genes were recognized manually by comparison.
The genomes were compared with T. trichiura and Trichuris sp. Sequences—T. trichiura from humans (unknown geographical location) (AP017704), T. trichiura from humans in China (NC_017750), T. trichiura from humans in China (GU385218), T. trichiura from humans in Uganda (KT449826), T. trichiura from P. anubis in USA (KT449825), T. trichiura from P. hamadryas in Denmark (KT449824) and Trichuris sp. from T. francoisi in China (KC461179). All protein-coding genes (PCGs) and ribosomal RNA genes (rRNAs) sequences were individually extracted, and a nucleotide data set was generated by concatenated sequences (all PCGs and rRNAs genes). This data set was used to estimate genetic distances using MEGA X v.10.1.8 (Penn State, PA, USA) [27]. The genomes of Trichuris from humans and NHP were used to calculate the < Nucleotide diversity (π) using a sliding window of 100 bp with 25 bp steps implemented in DnaSP v.6 [28].
2.5. Phylogenetic Analysis
For the phylogenetic analyses, two different data sets were generated. The first included nucleotides sequences, with the 13 PCGs (cox1–3, nad1–6, nad4L, atp6, atp8 and cob) and the two rRNAs (rrnL and rrnS). The data set was aligned using MEGA X v.10.1.8 [27] and concatenated for Trichuris sp. and T. trichiura species from baboons and humans. We used as an outgroup Trichinella pseudospiralis (Table 1).
Another data set was generated using amino acid sequences inferred from the 12 PCGs. The sequences were aligned using MEGA X v.10.1.8 [27] and then concatenated excluding atp8 (because it is not present in the mitochondrial genome of all the species of nematodes except in Trichinella and Trichuris species). The data set, using the sequences previously used and all the sequences available of the complete mt genome of Trichuris spp. and with those of 9 other enoplid nematodes, using Brugia malayi and Ascaris suum as the outgroups, was generated (Table 2). The ambiguous regions of the alignment were excluded using Gblocks Server v.0.91b (http://phylogeny.lirmm.fr/phylo_cgi/one_task.cgi?task_type=gblocks (accessed on 1 February 2021)) with the default settings being used to select the option of less strict conservation of flanking positions [29,30].
For phylogenetic re-constructions we used three methods—Maximum Likelihood (ML), Maximum Parsimony (MP) and Bayesian Inferences (BI). The ML tree was generated using PHYML package [31,32], and for the MP tree we used MEGA X v.10.1.8 [27] and for BI we used MrBayes v.3.2.6. [33]. To resolve the best-fit substitution model for the nucleotide data set we employed jModelTest [34] and ProtTest 3.4 for the amino acid data set. Models of evolution were defined according to the Akaike Information Criterion [35,36]. For the nucleotide data set, GTR + I + G model, with rate variation along the length of the alignment (+ G) and allowing for a proportion of invariant sites (+ I) was selected, and for the amino acid data set, the MtArt + I + G + F model, with residue frequencies estimated from the data (+ F) was chosen. Support for the topology was examined using bootstrapping (heuristic option) [37] over 1000 replications to assess the relative reliability of clades. The commands used in MrBayes for BI were nst = mixed. The standard deviation of split frequencies was used to determine whether the number of generations completed was sufficient; the chain was sampled every 500 generations and each dataset was run for 10 million generations. Trees from the first million generations were discarded based on an assessment of convergence. Burn-in was determined empirically by examination of the log likelihood values of the chains. The Bayesian posterior probabilities (BPPs) comprise the percentage converted.
3. Results
3.1. Annotation and Features of Mitochondrial Genomes
The complete mtDNA sequences of the primate worms TMF31, TMM5 and TPM1 were 14,091, 14,047 and 14,089 bp in length, respectively (GenBank accession nos. MW448470-2) (Figure 1). The mt genomes contained 37 genes—13 PCGs (cox1–3, nad1–6, nad4L, atp6, atp8 and cob), 22 transfer RNA genes (tRNAs), and two rRNAs (rrnL and rrnS) (Table 3). All genes are transcribed from the heavy strand, except four PCGs (nad2, nad4, nad4L and nad5) and 10 tRNA (tRNA-Met, tRNA-Phe, tRNA-His, tRNA-Arg, tRNA-Pro, tRNA-Trp, tRNA-Ile, tRNA-Gly, tRNA-Cys, and tRNA-Tyr) that are transcribed from the light strand.
The mt genomes of Trichuris sp. contain an AT-rich region consisting of two non-coding regions (NCRs), including a long non-coding region (NCR-L) and short non-coding region (NCR-S). The nucleotide composition (%) is summarized in Table S3. The content of A + T is 69.4%, 68% and 69.3% for TMH31, TMM5 and TPM1, respectively.
Between TMF31 and TPM1 genomes there were slight differences. The sequences were identical in terms of all initiation and termination codons and gene lengths, except for tRNA-ser (S2), which presented one nucleotide more in TPM1. In addition, between TMF31 and TMM5, 22 genes of 37 were different in length.
The start/stop codons for some PCGs differed between TMM5 and the genomes previously cited (TMF31 and TPM1), which were similar. Codon usage analyses of mt genomes showed that three start and seven termination codons were different (Table 3). For instance, the starting codon for TMF31 and TPM1 is ATA for the nad1 gene, while it reads ATG in the TMM5 genome; for the nad4 gene, the starting codon is ATG in TMF31 and TPM1, while being ATA in TMM5, and in the atp8 gene, ATT is the start codon in TMF31 and TPM1, while it reads ATA in TMM5. In the cox1 gene, TAA is the termination codon in TMF31 and TPM1, whereas it is TAG in TMM5; in cox2 gene, TAG is the stop codon in TMF31 and TPM1, while being TAA in TMM5; in nad1, TAG is the stop codon in TMF31 and TPM1, but is TAA in TMM5; in nad2 gene, TAA is the stop codon in TMF31 and TPM1, and is TAG in TMM5; the stop codon in the nad5 gene is TAG for TMF31 and TPM1 and TAA for TMM5; in the nad4L and nad6 genes, TAA is the stop codon in TMF31 and TPM1 while it reads TAG in TMM5, and in cox3 gene TAA is the stop codon for TMF31 and TPM1 and TAG for TMM5. There are overlaps between rrnL and atp6 in TMF31, between rrnL, atp6 and cox3 in TMM5, and between rrnL and atp6 in TPM1.
3.2. Comparative Sequence Analyses
Genetic distances between worms for individual PCGs and rRNAs genes are found in Table S2. The genetic distances between the mt genomes of Trichuris spp. in primates and humans are given in Table 4. The gene with highest genetic variation was the atp8 gene, and the most conserved was the rrnS gene, however between all PCGs, cox1 was the most conserved gene. Among the mt genomes obtained in this study, nucleotide and amino acid differences between TMF31 and TPM1 were 0.25% and 0.41%, and between TMF31 and TMM5 they were 18.7% and 14.5%, respectively. Within all sequences of Trichuris spp. studied, the mt genome of T. francoisi was the most variable (with a nucleotide difference of 27.1–28.6% and an amino acid difference of 26.8–28.2%) relative to the other worms from baboons and humans.
Among the Trichuris genome dataset, the nucleotide diversity was analyzed using the sliding window approach for the 13 PCGs and the two rRNA genes. The number of polymorphic sites was 5108 and the nucleotide diversity was 0.166 (Figure 2). The genes with lowest nucleotide diversity were rRNAs (rrnL and rrnS) and cox1.
3.3. Phylogenetic Analyses
The phylogenetics analyses of nucleotide sequence datasets with partial genome (the 13 PCGs (cox1–3, nad1–6, nad4L, atp6, atp8 and cytb) and the two rRNAs (rrnL and rrnS)) and with mt genome complete datasets reflecting similar tree topologies by the three methods studied (ML, MP and BI) (Figure 3). Within Trichuris populations from humans and NHPs, Trichuris sp. from T. francoisi appeared separated of the other sequences. Within this last group, there are three main clades. The first clade (clade 1) is composed of sequences of Trichuris sp. from H. sapiens and P. anubis from Asia, Japan and USA, the clade 2 is composed of a sequence of Trichuris sp. from M. sylvanus, and the clade 3 corresponded with T. trichiura from humans and NHPs (M. sylvanus, P. papio and P. hamadryas) from Europe and Africa.
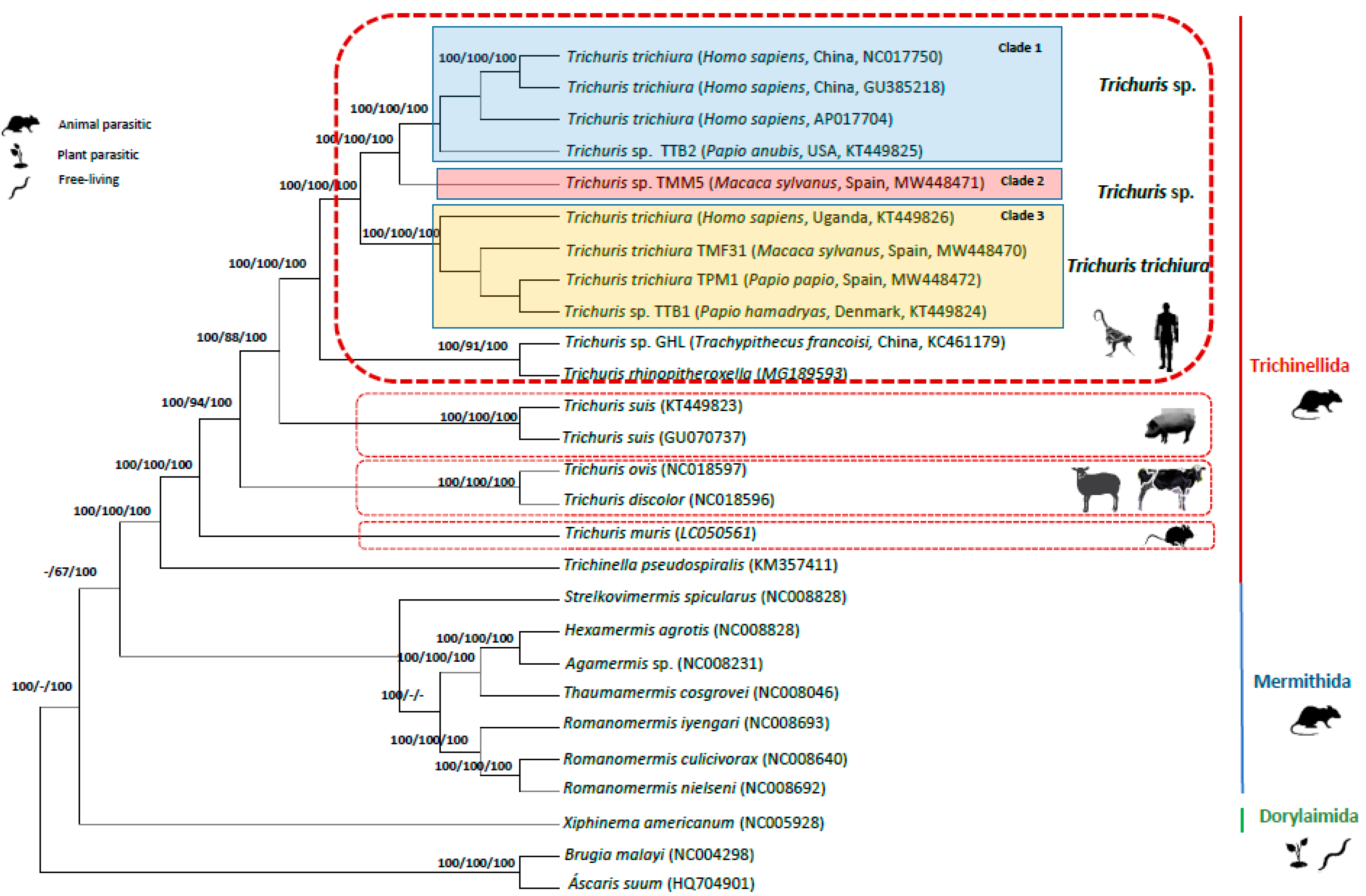
Phylogenetic trees inferred from a concatenated amino acid sequence dataset of 12 PCGs among selected enoplid nematodes, using the chromadorean nematode, B. malayi (NC_004298) and A. suum (HQ704901) as outgroups, revealed congruence with phylogenetics analyses of nucleotide sequence datasets with the 13 PCGs and the two rRNAs (rrnL and rrnS), and complete mt genomes. Thus, phylogenetics analyses of all 12 genes showed a general pattern with very strong support for clades near the terminals of the tree (Figure 4). Thus, clade 1, clade 2 and clade 3 for Trichuris spp. from humans and NHPs, and the monophyly of the genus Trichuris, are very strongly supported (100% ML BV, 100% MP BV and 100% BPP). There are no marked differences in support values between the 12 gene datasets analyzed using nucleotide versus amino acid sequences.
The phylogenetic analyses reflected the clear distinctiveness between T. trichiura and Trichuris sp. from humans and NHPs with respect to T. suis from suids, T. ovis and T. discolor from herbivorous and T. muris from rodents and grouping these members of Trichuris with T. pseudospiralis (order Trichinellida) with absolute support (100% ML BV, MP BV 100% and 100% BPP), but excluding the members of Dorylaimida and Mermithida (Figure 4).
4. Discussion
Previous studies have reported the hypothesis that a complex whipworm species exists in primates, suggesting that different Trichuris species infect primates and humans [3,19,23,38,39,40,41].
In the present study, we resolved the complete mt genome sequences of two haplotypes of Trichuris isolated from M. sylvanus and one complete mt genome of Trichuris from P. papio. The size of the complete mt genomes were within the range reported for the Trichuridae family (ranging from 13,904 bp (T. discolor) to 14,521 bp (T. suis)) [3,16,17,19,22]. The complete mtDNA is a circular molecule and encodes 37 genes—13 PCGs (atp6, atp8, cox1–3, cob, nad1–6 and nad4L), two rRNAs (rrnS and rrnL), and 22 for tRNAs.
The initiation codons (ATG, ATA and ATT) agreed with those reported in the pig-derived and human-derived Trichuris. [16,18,20], while T. ovis and T. discolor also employed TTG [3,16,19], and T. skrjabini sequence used only two start codons (ATG and ATA) [22]. Two termination codons (TAA and TAG) were used as stop codons in all the four species of Trichuridae family (T. suis, T. trichiura, T. ovis and T. discolor) except T. skrjabini, which possesses another stop codon, TGA [3,16,17,19,22]. The sequences of this study were in accordance with the most related species (primates derived Trichuris) [3,19].
The complete mt genomes of one of the haplotypes of Trichuris from macaques (TMF31) and that from baboon (TPM1) were genetically similar, with a difference in nucleotide and amino acid sequence of 0.25% and 0.41%, and having 14,091 and 14,098 bp, respectively, and both genomes being similar with that of T. trichiura from humans from Uganda, which was 14,079 bp in length, and with TTB1 (13,984 bp in length) from Trichuris sp. from P. hamadryas [19], with a sequence variation of 0.28–0.47%. In contrast, the complete mt genome of the other haplotype from macaques (TMM5) was 14,047 bp, like human T. trichiura from China that was 14,046 bp in length [3], showing a nucleotide difference of 15.9%. Moreover, the mt genome of Trichuris sp. from P. anubis (USA) was 14,009 bp [19], with a nucleotide difference to TMM5 of 15.8%. A substantial level of nucleotide difference was detected between TMF31 (Clade 3) with respect to Trichuris sp. included in clades 1 and 2 (15.8–18.8%). Comparison of the nucleotide and amino acid sequences between TMF31 and TMM5 was 18.7% and 14.5%, respectively. The sequence variation detected in the 13 PCGs between TMF31 and TMM5 was 20.3% (nucleotide sequence) and 34.4% (amino acid sequence). The percentage of dissimilarity observed between the different sequences corresponded to previously cited ranges within different nematode species. Thus, Hawash et al. [19] suggested the presence of two Trichuris species with a difference in nucleotide and amino acid sequences of around 18.8% and 14.6%, respectively, of Trichuris from Ugandan humans and Chinese humans. Furthermore, T. suis and T. trichiura presented an amino acid difference of 33.3–39.2% [19], T. trichiura from human and Trichuris sp. from leaf monkey was 29.4% [17], T. ovis and T. discolor ranged from 11.0–33.9% [16], 11.7% between Wucheria bancrofti and B. malayi [42], 10.3% between Chabertia ovina and C. erschowi [43], 4–18% between different Trichinella spp. [18] and 4.12% between Ancylostoma duodenale and Ancylostoma caninum [44]. Blouin [45] indicated that the difference in mt genome sequences between closely related species was normally 10–20%. Thus, the molecular evidence presented in the present manuscript supports the hypothesis that the haplotypes TMF31 and TMM5 of Trichuris from M. sylvanus display differences in amino acid and nucleotide sequences that are within the range of those previously reported by different authors to consider them as different species.
On the other hand, the complete mt genomes of Trichuris spp. from different hosts were evaluated to clarify the evolutionary relationships. Considering that complete mt genome sequences are maternally inherited and mutate at a rapid rate related to nuclear genes, theses markers could prove useful for evolutionary and phylogenetic analyses [46].
Phylogenetics trees inferred from both datasets (sequences dataset of 12 PCGs among enoplid nematodes and nucleotide sequence datasets with the 13 PCGs and the two rRNAs (rrnL and rrnS)) showed congruence with a high support for the differentiation between Trichuris spp. and the different clades within Trichuris populations parasitizing humans and NHPs (clade 1, clade 2 and clade 3). Furthermore, the monophyly of the genus Trichuris was strongly supported and grouping these species of Trichuris (traditionally named as Trichocephalida) with T. pseuospirallis (Trichinellida,) with the exclusion of the members of the Dorylaimida and Mermithida (Figure 4). Similar results were reported by Liu et al. [3,16] who characterized the complete mitochondrial genomes of several whipworms and compared them with other enoplid nematodes.
The atp8 gene is present in mitogenomes of the Order Trichinellida (Trichuris and Trichinella species), but it is usually absent in mitogenomes of members of the phylum Nematoda [3,13,15,16,17,18,19]. These authors reported the lack of atp8 in the mtDNA of Chromadorean nematodes (traditionally named as Class Secernentea) and it could be derived, within that lineage, after the divergence of Chromadorean nematodes from other groups. Furthermore, phylogenetic inferences confirm the clear relationships between Trichinellida and Mermithida correlating both groups with animal parasites of nematodes separated of Dorylaimida, corresponding to free-living nematodes and plant parasitic nematodes [47].
The phylogenetic study also supported the evolution of the different Trichuris species showing T. trichiura (clade 3), to be closer to Trichuris spp. (clade 1 and clade 2) parasitizing humans and NHPs. As reported by previous authors [19,23,38,39,41,48], Trichuris populations parasitizing humans and NHPs showed a species complex in primates. In addition, the high difference in nucleotide and amino acid sequences between T. trichiura (clade 3) and the minority population of Trichuris sp. from M. sylvanus (clade 2) and Trichuris sp. from humans and NHPs from China, Japan, and USA (clade 1) was confirmed in the phylogenetic trees with strong support for the different clades. Thus, based on our results, we suggest the existence of two cryptic species parasitizing M. sylvanus. Trichuris genus is a likely candidate to containing cryptic species as it has a wide geographical distribution and infects several host species [49]. As revealed by recent studies, there is more than one taxon capable of infecting humans and other primates, including individuals in captivity, suggesting that T. trichiura should be considered a complex species that includes different cryptic units [19]. In addition, and based on morphobiometric and molecular parameters, new species of Trichuris have been described in primates, such as T. rhinopiptheroxella [20], that was found in the golden snub-nosed monkey (Rhinopithecus roxellana), Trichuris colobae from Colobus guereza kikuyensis [50], and Trichuris ursinus from Papio ursinus [51]. As a result, it has been confirmed that T. trichiura and other Trichuris species are present in humans and NHPs.
This fact has two main epidemiological repercussions—(i) the zoonotic potential of Trichuris spp. of NHPs for humans. This is particularly important when humans and NHPs are living in proximity, as is becoming increasingly common with human encroachment into habitats where NHPs accessing gardens and farms in search of food and has significant implications for both human health and wildlife conservation [52]. This implies that in communities where access to NHPs is common, simple public health measures should be encouraged, including thorough handwashing with soap (particularly for children) and rising and cooking of vegetables, and (ii) the presence of different cryptic species might also be very important for implementation of appropriate control strategies. Different control strategies for Opistorchirs viverrini have been identified due to the existence of different cryptic species based on the different fecundity as measured by eggs/g/worm [53].
5. Conclusions
In the present study, based on complete mt genome analyses, the molecular data suggested two distinct species in whipworms isolated from M. sylvanus. The whipworms from P. papio, TPM1, and TMF31 from M. sylvanus are T. trichiura since the sequences were within the majority clade, with sequences from Ugandan humans and P. hamadryas (Europe)-derived Trichuris. Further, we suggested the existence of two cryptic species parasitizing M. sylvanus. Moreover, a major source of mitochondrial markers is given and may be used as the basis of subsequent epidemiological research, to clarify appropriate control measures and transmission routes.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-1729/11/2/126/s1, Table S1: PCR mix, primers and conditions used for each molecular marker sequenced in the present study, Table S2: Pairwise nucleotide and amino acid distances for the different 13 PCGs and RNAs (rrnS and rrnL) genes for Trichuris species from different primates and human hosts. Nucleotide genetics distances are given below the diagonal and amino acid genetic distances above the diagonal, Table S3: Nucleotide composition (%) of the mt genomes studied.
Author Contributions
Conceptualization, C.C. and R.C.; methodology, J.R. and R.C.; software, J.R.; validation, C.C., J.R. and R.C.; formal analyses, J.R. and R.C.; investigation, C.C., J.R. and C.R.; writing—original draft preparation, C.C., J.R. and R.C.; writing—review and editing, C.C., J.R. and R.C.; supervision, C.C.; project administration, C.C.; funding acquisition, C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by a grant from the Ministry of Economy, Industry and Competitiveness (CGL2017-83057), which included FEDER/Ministry of Science, Innovation and Universities – State Research Agency/ CGL2017-83057, the Junta de Andalucía (BIO-338) and a grant from the V and VI Plan Propio de Investigación of the University of Seville, Spain.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in GenBank database (GenBank accession nos. MW448470-2).
Acknowledgments
We thank Ruiz de la Haba for his technical scientific advice, and Zoo Castellar (Cádiz, Spain) for providing samples of Trichuris spp. from M. sylvanus which naturally died and Parque Natural de Cabárceno (Cantabria, Spain) for providing samples of T. trichiura from P. papio.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/soil-transmitted-helminth-infections. (accessed on 2 March 2020).
- Mogaji, H.O.; Dedeke, G.A.; Bada, B.S.; Bankole, S.; Adeniji, A.; Fagbenro, M.T.; Omitola, O.O.; Oluwole, A.S.; Odoemene, N.S.; Abe, E.M.; et al. Distribution of ascariasis, trichuriasis and hookworm infections in Ogun State, Southwestern Nigeria. PLoS ONE 2020, 15, e0233423. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Gasser, R.B.; Su, A.; Nejsum, P.; Peng, L.; Lin, R.Q.; Li, M.W.; Xu, M.J.; Zhu, X.Q. Clear genetic distinctiveness between human-and pig-derived Trichuris based on analysis of mitochondrial datasets. PLoS Negl. Trop. Dis. 2012, 6, e1539. [Google Scholar] [CrossRef] [Green Version]
- Summers, R.W.; Elliott, D.E.; Urban, J.F., Jr.; Thompson, R.; Weinstock, J.V. Trichuris suis therapy in Crohn’s disease. Gut 2005, 54, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, R.W.; Elliott, D.E.; Urban, J.F., Jr.; Thompson, R.A.; Weinstock, J.V. Trichuris suis therapy for active ulcerative colitis: A randomized controlled trial. Gastroenterology 2005, 128, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, R.W.; Elliott, D.E.; Qadir, K.; Urban, J.F., Jr.; Thompson, R.; Weinstock, J.V. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am. J. Gastroenterol. 2003, 98, 2034–2041. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Arnved, J.; Rønborg, S.; Wohlfahrt, J.; Poulsen, L.K.; Westergaard, T.; Petersen, H.W.; Kristensen, B.; Thamsborg, S.; Roepstorff, A.; et al. Trichuris suis ova therapy for allergic rhinitis: A randomized, double-blind, placebo-controlled clinical trial. J. Allergy Clin. Immunol. 2010, 125, 123–130.e3. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Hamelmann, E.; Lucius, R.; Hartmann, S. Looking into the future of Trichuris suis therapy. J. Allergy Clin. Immunol. 2010, 125, 767–769. [Google Scholar] [CrossRef]
- Cantacessi, C.; Young, N.D.; Nejsum, P.; Jex, A.R.; Campbell, B.E.; Hall, R.S.; Thamsborg, S.M.; Scheerlinck, J.P.; Gasser, R.B. The transcriptome of Trichuris suis—First molecular insights into a parasite with curative properties for key immune diseases of humans. PLoS ONE 2011, 6, e23590. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.; Fried, B. The use of Trichuris suis and other helminth therapies to treat Crohn’s disease. Parasitol. Res. 2007, 100, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Elliott, D.E.; Weinstock, J.; Summers, R.W.; Landry-Wheeler, A.; Silver, N.; Harnett, M.D.; Hanauer, S.B. Randomised clinical trial: The safety and tolerability of Trichuris suis ova in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2013, 38, 255–263. [Google Scholar] [CrossRef]
- Hiemstra, I.H.; Klaver, E.J.; Vrijland, K.; Kringel, H.; Andreasen, A.; Bouma, G.; Kraal, G.; van Die, I.; den Haan, J.M. Excreted/secreted Trichuris suis products reduce barrier function and suppress inflammatory cytokine production of intestinal epithelial cells. Mol. Immunol. 2014, 60, 1–7. [Google Scholar] [CrossRef]
- Kern, E.M.A.; Kim, T.; Park, J.-K. The Mitochondrial Genome in Nematode Phylogenetics. Front. Ecol. Evol. 2020, 8, 250. [Google Scholar] [CrossRef]
- Egger, B.; Bachmann, L.; Fromm, B. Atp8 is in the ground pattern of flatworm mitochondrial genomes. BMC Genomics 2017, 18, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrov, D.V.; Brown, W.M. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics 2001, 157, 621–637. [Google Scholar]
- Liu, G.H.; Wang, Y.; Xu, M.J.; Zhou, D.H.; Ye, Y.G.; Li, J.Y.; Li, J.; Song, H.; Lin, R.; Zhu, X.Q. Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae). Infect. Genet. Evol. 2012, 12, 1635–1641. [Google Scholar] [CrossRef]
- Liu, G.H.; Gasser, R.B.; Nejsum, P.; Wang, Y.; Chen, Q.; Song, H.Q.; Zhu, X.Q. Mitochondrial and nuclear ribosomal DNA evidence supports the existence of a new Trichuris species in the endangered françois’ leaf-monkey. PLoS ONE 2013, 8, e66249. [Google Scholar] [CrossRef] [Green Version]
- Mohandas, N.; Pozio, E.; La Rosa, G.; Korhonen, P.K.; Young, N.D.; Koehler, A.V.; Hall, R.S.; Sternberg, P.W.; Boag, P.R.; Jex, A.R.; et al. Mitochondrial genomes of Trichinella species and genotypes—A basis for diagnosis, and systematic and epidemiological explorations. Int. J. Parasitol. 2014, 44, 1073–1080. [Google Scholar] [CrossRef]
- Hawash, M.B.; Andersen, L.O.; Gasser, R.B.; Stensvold, C.; Nejsum, P. Mitochondrial genome analyses suggest multiple Trichuris species in humans, baboons, and pigs from different geographical regions. PLoS Negl. Trop. Dis. 2015, 9, e0004059. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Song, L.; Zhu, L.; Chen, M.; Ren, G.; Liu, G.H.; Zhao, G.H. Morphological and molecular confirmation of the validity of Trichuris rhinopiptheroxella in the endangered golden snub-nosed monkey (Rhinopithecus roxellana). J. Helminthol. 2019, 93, 601–607. [Google Scholar] [CrossRef]
- Kim, J.; Kern, E.; Kim, T.; Sim, M.; Kim, J.; Kim, Y.; Park, C.; Nadler, S.A.; Park, J.K. Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within Chromadorea. Mol. Phylogenet. Evol. 2017, 107, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.A.; Shabbir, M.A.B.; Xin, Y.; Ikram, M.; Hafeez, M.A.; Wang, C.; Zhang, T.; Zhou, C.; Yan, X.; Hassan, M.; et al. Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae). Genes 2019, 10, 438. [Google Scholar] [CrossRef] [Green Version]
- Rivero, J.; Cutillas, C.; Callejón, R. Trichuris trichiura (Linnaeus, 1771) from human and non-human primates: Morphology, biometry, host specificity, molecular characterization, and phylogeny. Front. Vet. Parasitol. (in press).
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phyl. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.; Chan, P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Cavallero, S.; De Liberato, C.; Friedrich, K.G.; Di Cave, D.; Masella, V.; D’Amelio, S.; Berrilli, F. Genetic heterogeneity and phylogeny of Trichuris spp. from captive non-human primates based on ribosomal DNA sequence data. Infect. Genet. Evol. 2015, 34, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Cavallero, S.; Nejsum, P.; Cutillas, C.; Callejón, R.; Doležalová, J.; Modrý, D.; D’Amelio, S. Insights into the molecular systematics of Trichuris infecting captive primates based on mitochondrial DNA analysis. Vet. Parasitol. 2019, 272, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, B.; Hoberg, E.P.; Li, M.; Zhou, X.; Gu, X.; Lai, W.; Peng, X.; Yang, G. Genetic characterisation and phylogenetic status of whipworms (Trichuris spp.) from captive non-human primates in China, determined by nuclear and mitochondrial sequencing. Parasit. Vectors 2018, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Rivero, J.; García-Sánchez, Á.M.; Zurita, A.; Cutillas, C.; Callejón, R. Trichuris trichiura isolated from Macaca sylvanus: Morphological, biometrical, and molecular study. BMC Vet. Res. 2020, 16, 445. [Google Scholar] [CrossRef]
- Ramesh, A.; Small, S.T.; Kloos, Z.A.; Kazura, J.W.; Nutman, T.B.; Serre, D.; Zimmerman, P.A. The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 2012, 183, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Zhao, L.; Song, H.Q.; Zhao, G.H.; Cai, J.Z.; Zhao, Q.; Zhu, X. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasit. Vectors 2014, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Jex, A.R.; Waeschenbach, A.; Hu, M.; van Wyk, J.A.; Beveridge, I.; Littlewood, D.T.; Gasser, R.B. The mitochondrial genomes of Ancylostoma caninum and Bunostomum phlebotomum—two hookworms of animal health and zoonotic importance. BMC Genomics 2009, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blouin, M.S. Molecular prospecting for cryptic species of nematodes: Mitochondrial DNA versus internal transcribed spacer. Int. J. Parasitol. 2002, 32, 527–531. [Google Scholar] [CrossRef]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 2002, 32, 145–158. [Google Scholar] [CrossRef]
- Wasmuth, J.; Schmid, R.; Hedley, A.; Blaxter, M. On the extent and origins of genic novelty in the phylum Nematoda. PLOS Negl. Trop. Dis. 2008, 2, e258. [Google Scholar] [CrossRef]
- Ravasi, D.F.; O’Riain, M.J.; Davids, F.; Illing, N. Phylogenetic evidence that two distinct Trichuris genotypes infect both humans and non-human primates. PLoS ONE 2012, 7, e44187. [Google Scholar] [CrossRef] [Green Version]
- Nadler, S.A.; DE León, G.P. Integrating molecular and morphological approaches for characterizing parasite cryptic species: Implications for parasitology. Parasitology 2011, 138, 1688–1709. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, C.; De Rojas, M.; Zurita, A.; Oliveros, R.; Callejón, R. Trichuris colobae n. sp. (Nematoda: Trichuridae), a new species of Trichuris from Colobus guereza kikuyensis. Parasitol. Res. 2014, 113, 2725–2732. [Google Scholar] [CrossRef] [PubMed]
- Callejón, R.; Halajian, A.; Cutillas, C. Description of a new species, Trichuris ursinus n. sp. (Nematoda: Trichuridae) from Papio ursinus Keer, 1792 from South Africa. Infect. Genet. Evol. 2017, 51, 182–193. [Google Scholar] [CrossRef]
- Betson, M.; Søe, M.; Nejsum, P. Human trichuriasis: Whipworm genetics, phylogeny, transmission and future research directions. Curr. Trop. Med. Rep. 2015, 2, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Saijuntha, W.; Sithithaworn, P.; Wongkham, S.; Laha, T.; Pipitgool, V.; Tesana, S.; Chilton, N.B.; Petney, T.N.; Andrews, R.H. Evidence of a species complex within the food-borne trematode Opisthorchis viverrini and possible co-evolution with their first intermediate hosts. Int. J. Parasitol. 2007, 37, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mitochondrial genome structure of Trichuris trichiura from Barbary macaque (TMF31) and from Guinea baboon (TPM1), and Trichuris sp. from Barbary macaque (TMM5). Genes were represented to standard nomenclature, but tRNAs were represented using one-letter amino acid codes, with numbers differentiating each of the two leucine- and serine-specifying tRNAs. NCR-L refers to large non-coding region and NCR-S to small non-coding region.
Figure 1.
Mitochondrial genome structure of Trichuris trichiura from Barbary macaque (TMF31) and from Guinea baboon (TPM1), and Trichuris sp. from Barbary macaque (TMM5). Genes were represented to standard nomenclature, but tRNAs were represented using one-letter amino acid codes, with numbers differentiating each of the two leucine- and serine-specifying tRNAs. NCR-L refers to large non-coding region and NCR-S to small non-coding region.
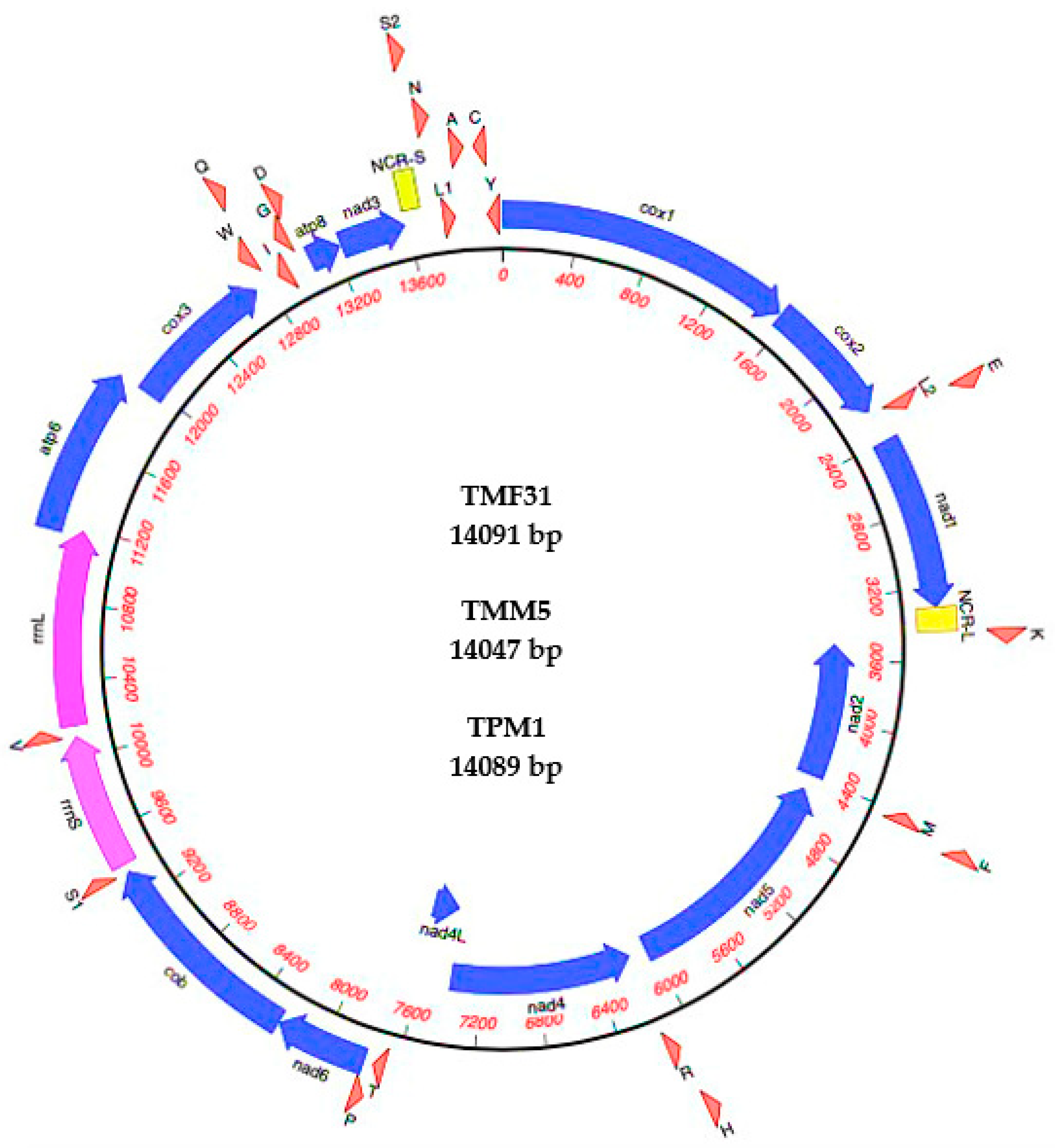
Figure 2.
Nucleotide diversity (π) for all protein-coding genes (PCGs) and ribosomal rRNA (rrnS and rrnL) measured using a sliding window of 100 bp with 25 bp steps. The aligned dataset for Trichuris in primates (baboons, humans and Francois’ leaf monkey).
Figure 2.
Nucleotide diversity (π) for all protein-coding genes (PCGs) and ribosomal rRNA (rrnS and rrnL) measured using a sliding window of 100 bp with 25 bp steps. The aligned dataset for Trichuris in primates (baboons, humans and Francois’ leaf monkey).

Figure 3.
Phylogenetic tree based on concatenated nucleotide sequences of 13 PCGs and two rRNA of Trichuris from baboons, humans and françois’ leaf monkey using Trichinella pseudospiralis as an outgroup, inferred using Bayesian Inference (BI). Maximum Likelihood (ML) bootstrap values of clades are listed first, followed by Maximum Parsimony (MP) and by Bayesian Posterior Probabilities (BPP), for clade frequencies exceeding 60%.
Figure 3.
Phylogenetic tree based on concatenated nucleotide sequences of 13 PCGs and two rRNA of Trichuris from baboons, humans and françois’ leaf monkey using Trichinella pseudospiralis as an outgroup, inferred using Bayesian Inference (BI). Maximum Likelihood (ML) bootstrap values of clades are listed first, followed by Maximum Parsimony (MP) and by Bayesian Posterior Probabilities (BPP), for clade frequencies exceeding 60%.
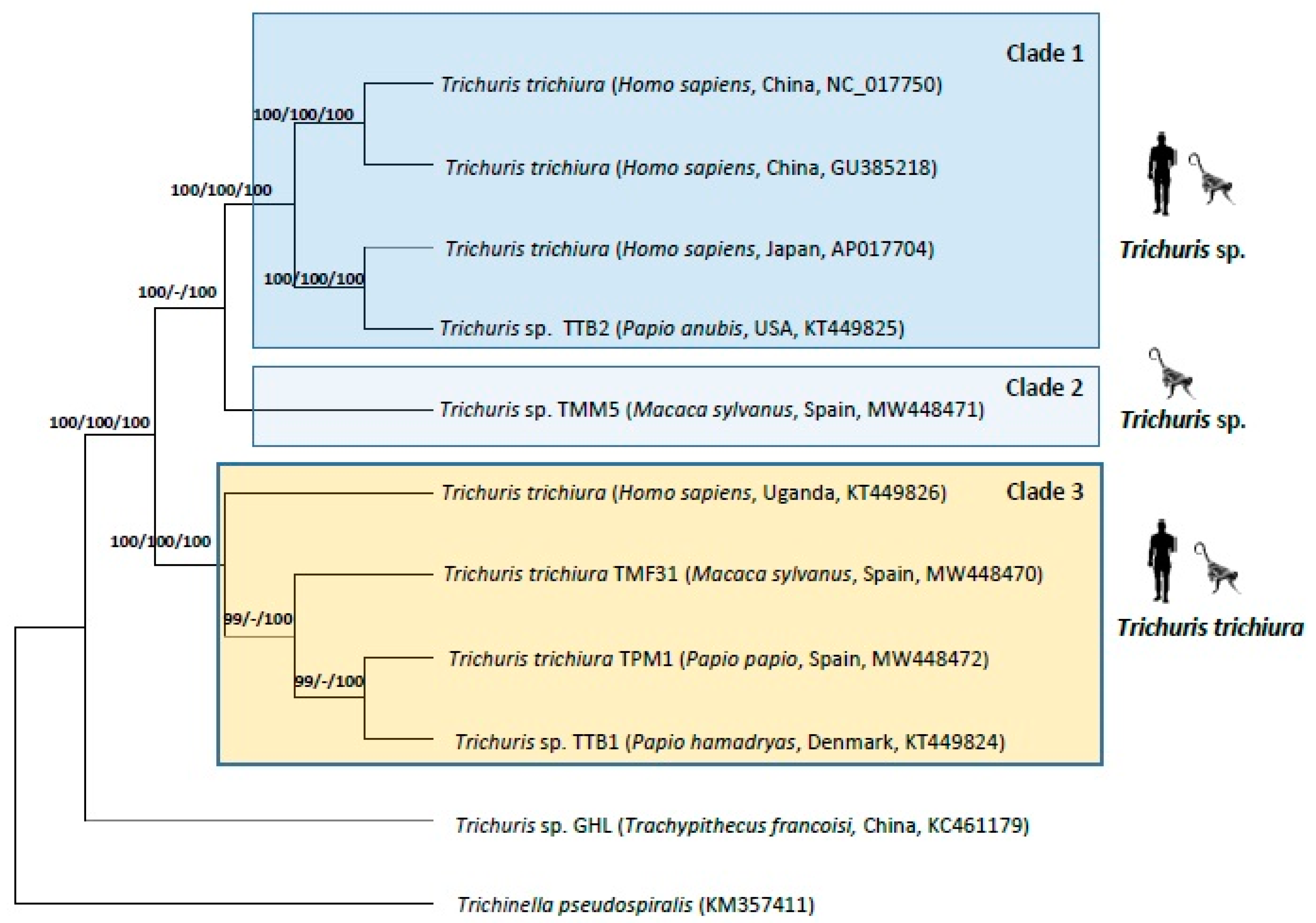
Figure 4.
Phylogenetic tree among enoplid nematodes based on concatenated amino acid sequences of 12 PCGs (except for atp8 gene) by Bayesian Inference (BI) using Brugia malayi and Ascaris suum as the outgroups. Maximum Likelihood (ML) bootstrap values of clades are listed first, followed by Maximum Parsimony (MP) and Bayesian Posterior Probabilities, for clade frequencies exceeding 60%.
Figure 4.
Phylogenetic tree among enoplid nematodes based on concatenated amino acid sequences of 12 PCGs (except for atp8 gene) by Bayesian Inference (BI) using Brugia malayi and Ascaris suum as the outgroups. Maximum Likelihood (ML) bootstrap values of clades are listed first, followed by Maximum Parsimony (MP) and Bayesian Posterior Probabilities, for clade frequencies exceeding 60%.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sequences analyzed in the first data set for phylogenetic analyses.
| Species | Host Species/Geographical Origin | GenBank Accession Number |
|---|---|---|
| Trichuris trichiura | Macaca sylvanus/Spain | MW448470 |
| Trichuris sp. | Macaca sylvanus/Spain | MW448471 |
| Trichuris trichiura | Papio papio/Spain | MW448472 |
| Trichuris trichiura | Homo sapiens/(unknown geographical location) | AP017704 |
| Trichuris trichiura | Homo sapiens/China | NC_017750 |
| Trichuris trichiura | Homo sapiens/China | GU385218 |
| Trichuris trichiura | Homo sapiens/Uganda | KT449826 |
| Trichuris sp. | Papio anubis/USA | KT449825 |
| Trichuris sp. | Papio hamadryas/Denmark | KT449824 |
| Trichuris sp. | Trachypithecus francoisi/China | KC461179 |
| 1Trichinella pseudospiralis | Coragypus atratus/USA | KM357411 |
1 Used as an outgroup.
Table 2.
Sequences analyzed in the second data set for phylogenetic analyses.
| Species | Host Species/Geographical Origin | Order | GenBank Accession Number |
|---|---|---|---|
| Trichuris trichiura | Macaca sylvanus/Spain | Trichinellida | MW448470 |
| Trichuris sp. | Macaca sylvanus/Spain | Trichinellida | MW448471 |
| Trichuris trichiura | Papio papio/Spain | Trichinellida | MW448472 |
| Trichuris trichiura | Homo sapiens/(unknown geographical location) | Trichinellida | AP017704 |
| Trichuris trichiura | Homo sapiens/China | Trichinellida | NC_017750 |
| Trichuris trichiura | Homo sapiens/China | Trichinellida | GU385218 |
| Trichuris trichiura | Homo sapiens/Uganda | Trichinellida | KT449826 |
| Trichuris sp. | Papio anubis/USA | Trichinellida | KT449825 |
| Trichuris sp. | Papio hamadryas/Denmark | Trichinellida | KT449824 |
| Trichuris sp. | Trachypithecus francoisi/China | Trichinellida | KC461179 |
| Trichuris rhinopiptheroxella | Rhinopithecus roxellana/China | Trichinellida | MG189593 |
| Trichuris ovis | Addax nasomaculatus/China | Trichinellida | NC_018597 |
| Trichuris discolor | Bos grunniens mutus/China | Trichinellida | NC_018596 |
| Trichuris muris | - /United Kingdom | Trichinellida | LC050561 |
| Trichuris suis | Sus scrofa/Uganda | Trichinellida | KT449823 |
| Trichuris suis | Sus scrofa/China | Trichinellida | GU070737 |
| Trichinella pseudospiralis | Coragypus atratus/USA | Trichinellida | KM357411 |
| Xiphinema americanum | Plant ectoparasite | Dorylaimida | NC_005928 |
| Hexamermis agrotis | - | Mermithida | NC_008828 |
| Agamermis sp. | - | Mermithida | NC_008231 |
| Romanomermis culicivorax | - | Mermithida | NC_008640 |
| Romanomermis iyengari | - | Mermithida | NC_008693 |
| Romanomermis nielseni | - | Mermithida | NC_008692 |
| Strelkovimermis spiculatus | - | Mermithida | NC_008047 |
| Thaumamermis cosgrovei | - | Mermithida | NC_008046 |
| 1Brugia malayi | - | Rhabditida | NC_004298 |
| 1Ascaris suum | - | Rhabditida | HQ704901 |
1 Used as an outgroup.
Table 3.
Mitochondrial genomes of Trichuris from Macaca sylvanus (TMF31, TMM5) and Papio papio (TPM1). Protein coding, transfer RNA (tRNA), and ribosomal RNA (rRNA) genes with lengths in nucleotides (nt) are given. The lengths are not identical, and differences are given in parentheses (TMM5/TPM1), likewise for the initiation and termination codons.
Table 3.
Mitochondrial genomes of Trichuris from Macaca sylvanus (TMF31, TMM5) and Papio papio (TPM1). Protein coding, transfer RNA (tRNA), and ribosomal RNA (rRNA) genes with lengths in nucleotides (nt) are given. The lengths are not identical, and differences are given in parentheses (TMM5/TPM1), likewise for the initiation and termination codons.
| Genes | Positions | Lengths | Codons | Strand | |||
|---|---|---|---|---|---|---|---|
| TMF31 | TMM5 | TPM1 | nt | Initiation | Termination | ||
| cox1 | 1–1545 | 1–1545 | 1–1545 | 1545 | ATG | TAA (TAG) | + |
| cox2 | 1558–2232 | 1556–2230 | 1558–2232 | 675 | ATG | TAG (TAA) | + |
| tRNA-leu (L2) | 2255–2317 | 2252–2311 | 2255–2317 | 63 (60) | + | ||
| tRNA-glu (E) | 2324–2384 | 2320–2377 | 2324–2384 | 61 (58) | + | ||
| nad1 | 2406–3305 | 2401–3300 | 2406–3305 | 900 | ATA (ATG) | TAG (TAA) | + |
| Non-coding region (NCR-L) | 3306–3435 | 3303–3442 | 3306–3430 | ||||
| tRNA-lys (K) | 3436–3501 | 3441–3502 | 3431–3496 | 66 (62) | + | ||
| nad2 | 3499–4395 | 3505–4401 | 3494–4390 | 897 | ATA | TAA (TAG) | − |
| tRNA-met (M) | 4396–4456 | 4402–4462 | 4391–4451 | 61 | − | ||
| tRNA-phe (F) | 4451–4507 | 4457–4513 | 4446–4502 | 57 | − | ||
| nad5 | 4499–6055 | 4520–6067 | 4494–6050 | 1557 (1553) | ATA | TAG (TAA) | − |
| tRNA-his (H) | 6049–6106 | 6065–6118 | 6044–6101 | 58 (54) | − | ||
| tRNA-arg (R) | 6108–6171 | 6115–6181 | 6103–6166 | 64 (67) | − | ||
| nad4 | 6176–7396 | 6183–7394 | 6171–7391 | 1221 (1212) | ATG (ATA) | TAA | − |
| nad4L | 7419–7631 | 7425–7673 | 7414–7626 | 213 (249) | ATA | TAA (TAG) | − |
| tRNA-thr (T) | 7672–7729 | 7679–7736 | 7667–7724 | 58 | + | ||
| tRNA-pro (P) | 7729–7787 | 7736–7795 | 7724–7782 | 59 (60) | − | ||
| nad6 | 7780–8256 | 7788–8264 | 7775–8251 | 477 | ATT | TAA (TAG) | + |
| cob | 8263–9369 | 8272–9378 | 8258–9364 | 1107 | ATG | TAG | + |
| tRNA-ser (S1) | 9368–9420 | 9377–9426 | 9363–9415 | 53 (50) | + | ||
| rrnS | 9413–10116 | 9419–10112 | 9408–10,111 | 704 (694) | + | ||
| tRNA-val (V) | 10,118–10,174 | 10,114–10,170 | 10,113–10,169 | 57 | + | ||
| rrnL | 10,176–11,184 | 10,170–11,180 | 10,171–11,179 | 1009 (1011) | + | ||
| atp6 | 11,155–11,967 | 11,151–11,990 | 11,150–11,962 | 813 (840) | ATG | TAA | + |
| cox3 | 11,973–12,746 | 11,965–12,738 | 11,968–12,741 | 774 | ATG | TAA (TAG) | + |
| tRNA-trp (W) | 12,759–12,821 | 12,743–12,805 | 12,754–12,816 | 63 | − | ||
| tRNA-gln (Q) | 12,825–12,880 | 12,807–12,862 | 12,820–12,875 | 56 | + | ||
| tRNA-Ile (I) | 12,883–12,943 | 12,864–12,925 | 12,878–12,938 | 61 (62) | − | ||
| tRNA-gly (G) | 12,957–13,013 | 12,934–12,989 | 12,952–13,008 | 57 (56) | − | ||
| tRNA-asp (D) | 13,020–13,077 | 12,996–13,060 | 13,015–13,072 | 58 (65) | + | ||
| atp8 | 13,066–13,233 | 13,042–13,209 | 13,061–13,228 | 168 | ATT (ATA) | TAG | + |
| nad3 | 13,243–13,584 | 13,219–13,560 | 13,238–13,579 | 342 | ATT | TAA | + |
| Non-coding region NCR-S) | 13,585–13,676 | 13,561–13,659 | 13,580–13,672 | ||||
| tRNA-ser (S2) | 13,677–13,726 | 13,660–13,709 | 13,673–13,723 | 50 (50/51) | + | ||
| tRNA-asn (N) | 13,727–13,781 | 13,710–13,763 | 13,724–13,778 | 55 (54) | + | ||
| tRNA-leu (L1) | 13,789–13,848 | 13,770–13,835 | 13,786–13,845 | 60 (66) | + | ||
| tRNA-ala (A) | 13,860–13,917 | 13,842–13,897 | 13,857–13,914 | 58 (56) | + | ||
| tRNA-cys (C) | 13,960–14,013 | 13,924–13,976 | 13,957–14,010 | 54 (53) | − | ||
| tRNA-tyr (Y) | 14,014–14,074 | 13,977–14,038 | 14,011–14,071 | 61 (62) | − | ||
| Total length | 14,091 | 14,047 | 14,089 | ||||
Table 4.
Pairwise genetic and protein distances between the different complete mt genomes for different Trichuris trichiura and Trichuris sp. in different primates and human hosts at different countries. The nucleotide distances are given below the diagonal and the amino acid distances above the diagonal.
Table 4.
Pairwise genetic and protein distances between the different complete mt genomes for different Trichuris trichiura and Trichuris sp. in different primates and human hosts at different countries. The nucleotide distances are given below the diagonal and the amino acid distances above the diagonal.
| TMF31 | TPM1 | TMM5 | AP017704 T. trichiura H. sapiens (Unknown Geographical Location) | NC_017750 T. trichiura H. sapiens China | GU385218 T. trichiura H. sapiens China | KT2449826 T. trichiura H. sapiens Uganda | KT449825 Trichuris sp. TTB2 P. anubis USA | KT449824 Trichuris sp. P. hamadryas Denmark | KC461179 Trichuris sp. GHL T. francoisi China | |
|---|---|---|---|---|---|---|---|---|---|---|
| TMF31 | 0.41 | 14.5 | 14.7 | 14.8 | 14.8 | 0.6 | 14.6 | 0.49 | 26.9 | |
| TPM1 | 0.25 | 14.6 | 14.8 | 14.9 | 14.9 | 0.7 | 14.6 | 0.34 | 26.8 | |
| TMM5 | 18.7 | 18.7 | 10.9 | 11.1 | 11.1 | 14.5 | 10.3 | 14.6 | 28.2 | |
| AP017704 T. trichiura H. sapiens (unknown geographical location) | 18.7 | 18.7 | 15.7 | 4.68 | 4.68 | 14.7 | 3.47 | 14.9 | 28.3 | |
| NC_017750 T. trichiura H. sapiens China | 18.6 | 18.6 | 15.9 | 6.51 | 0 | 14.8 | 4.66 | 14.9 | 27.8 | |
| GU385218 T. trichiura H. sapiens China | 18.6 | 18.6 | 15.9 | 6.51 | 0 | 14.8 | 4.66 | 14.9 | 27.8 | |
| KT2449826 T. trichiura H. sapiens Uganda | 0.4 | 0.47 | 18.8 | 18.7 | 18.6 | 18.6 | 14.5 | 0.78 | 26.8 | |
| KT449825 Trichuris sp. TTB2 P. anubis USA | 18.8 | 18.8 | 15.8 | 4.7 | 6.48 | 6.48 | 18.8 | 14.7 | 28.2 | |
| KT449824 Trichuris sp. P. hamadryas Denmark | 0.34 | 0.28 | 18.8 | 18.7 | 18.6 | 18.6 | 0.55 | 18.8 | 26.8 | |
| KC461179 Trichuris sp. GHL T. francoisi China | 27.1 | 27.1 | 28.4 | 28.3 | 28 | 28 | 27.1 | 28.6 | 27.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rivero, J.; Callejón, R.; Cutillas, C. Complete Mitochondrial Genome of Trichuristrichiura from Macaca sylvanus and Papio papio. Life 2021, 11, 126. https://doi.org/10.3390/life11020126
AMA Style
Rivero J, Callejón R, Cutillas C. Complete Mitochondrial Genome of Trichuristrichiura from Macaca sylvanus and Papio papio. Life. 2021; 11(2):126. https://doi.org/10.3390/life11020126
Chicago/Turabian StyleRivero, Julia, Rocío Callejón, and Cristina Cutillas. 2021. "Complete Mitochondrial Genome of Trichuristrichiura from Macaca sylvanus and Papio papio" Life 11, no. 2: 126. https://doi.org/10.3390/life11020126
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.