1. Introduction
More than a decade ago the innovative idea arose to use parasitic helminths with active immunomodulatory properties to treat autoimmune and inflammatory diseases. Considering the potential benefits and risks of applying a known infectious organism to patients suffering from inflammatory conditions, the porcine whipworm
Trichuris suis was preselected as a promising candidate for treatment [
1].
T. suis is the species most closely related to the human-infecting parasite
T. trichiura and oral administration of
T. suis eggs (
T. suis ova, TSO) typically results in a self-limiting colonization of the human gut [
2,
3]. In contrast to ascarids or hookworms, whipworms like
T. suis have a direct, strictly enteric life cycle. Following ingestion of infective eggs (TSO) containing stage 1 larvae (L1), these larvae hatch and invade the mucosa of the large intestine of the host. There,
Trichuris larvae undergo four moulting steps. As they reach the adult stage, the thinner anterior end of the worm is found burrowed into the epithelial cell layer, while the thicker posterior end protrudes freely into the intestinal lumen [
4]. In humans, the pig whipworm
T. suis typically does not develop into sexually mature adults. Thus, in order to maintain hatched
T. suis larvae in the non-natural human host, repeated treatments with TSO are required.
The potential of TSO-based therapies to treat autoimmune diseases has been tested in a variety of clinical studies (reviewed in [
5]) with some promising, but also various disappointing clinical outcomes. Notably, such alternative helminth-based treatment has received considerable attention in recent years as a potentially useful therapy for treatment of multiple sclerosis (MS). MS is a debilitating, demyelinating disease of the central nervous system, most commonly presented as relapsing-remitting multiple sclerosis (RRMS), characterized by transient episodes of neurological dysfunction, mostly in young females [
6,
7]. Similar to other autoimmune diseases, the inverse correlation of natural helminth infections and the prevalence of MS in helminth-endemic countries was an initial driver of the idea of helminth-based therapy as a novel treatment option [
8]. Correale and Farez have demonstrated that the administration of anthelmintic drugs led to an increase of MS disease activity, suggestive of active suppression of MS mediated by helminth parasites [
9]. Moreover, several studies using experimental autoimmune encephalomyelitis (EAE), the murine model for MS, have tested infective helminth eggs, larvae or the application of helminth-derived molecules in both prophylactic and therapeutic treatment settings, reporting an improved neuroinflammatory disease outcome (reviewed in [
10]).
Until now, four clinical phase 1 and phase 2 trials investigating treatment effects of either oral inoculation of TSO or intradermal application of
Necator americanus (hookworm) larvae in MS patients have been completed [
11,
12,
13,
14]. While all studies demonstrate the safety of controlled administration of both
T. suis and
N. americanus, none of them reported strong therapeutic efficacy, expressed in radiological or clinical disease activity [
11,
12,
13,
14]. Immunological results of the HINT-2 study demonstrated that TSO given at a dose of 2500 TSO every 2 weeks over 10 months resulted in mild eosinophilia and
T. suis-specific antibodies [
13]. Additionally, the study showed an increase of CD4
+CD127
−CD25
high regulatory T cells (Treg) in the circulation of TSO-treated patients, compared with placebo-treated MS patients. Live
T. suis larvae were also observed in one of two TSO-treated patients undergoing clinically indicated colonoscopy, indicating that
T. suis larvae colonized the host intestine and induced a specific immune response [
13]. Similarly, preliminary results from four patients with secondary progressive MS receiving TSO treatment for compassionate use indicated a slight downregulation of Th1-associated cytokines [
15].
Combined, these results prompted us to set up a small monocentric, prospective, randomized, placebo-controlled, double-blind, phase II study enrolling patients with RRMS and clinically isolated syndrome (CIS) to investigate in greater detail changes in different immune parameters, as well as the individual helminth-specific T cell signatures of MS patients receiving prolonged TSO treatment (TRIOMS) [
16].
2. Materials and Methods
2.1. Patient Recruitment, Study Design, and Treatment Allocation
Eleven patients diagnosed with relapsing-remitting multiple sclerosis (RRMS) according to the revised McDonald criteria [
17] (CIS) with clinical activity were recruited for experimental treatment with
T. suis ova (TRIOMS). This study was a monocentric, prospective, randomized, placebo-controlled, double-blind, phase II pilot study, conducted at the Department of Neurology and the NeuroCure Clinical Research Center of the Charité-Universitätsmedizin Berlin [
16] (ClinicalTrials.gov: NCT01413243). The study was approved by the local ethics committee and the German Federal Institute for Drugs and Medical Devices (BfArM).
Individuals were recruited between September 2012 and March 2015. Patients aged between 18 and 65 years, with a score of ≤4.0 on the Expanded Disability Status Scale (EDSS) [
18], with disease activity on brain MRI, and without any assigned standard immunomodulatory therapies for at least 3 months prior to the study, were randomized 1:1 to receive orally either 2500
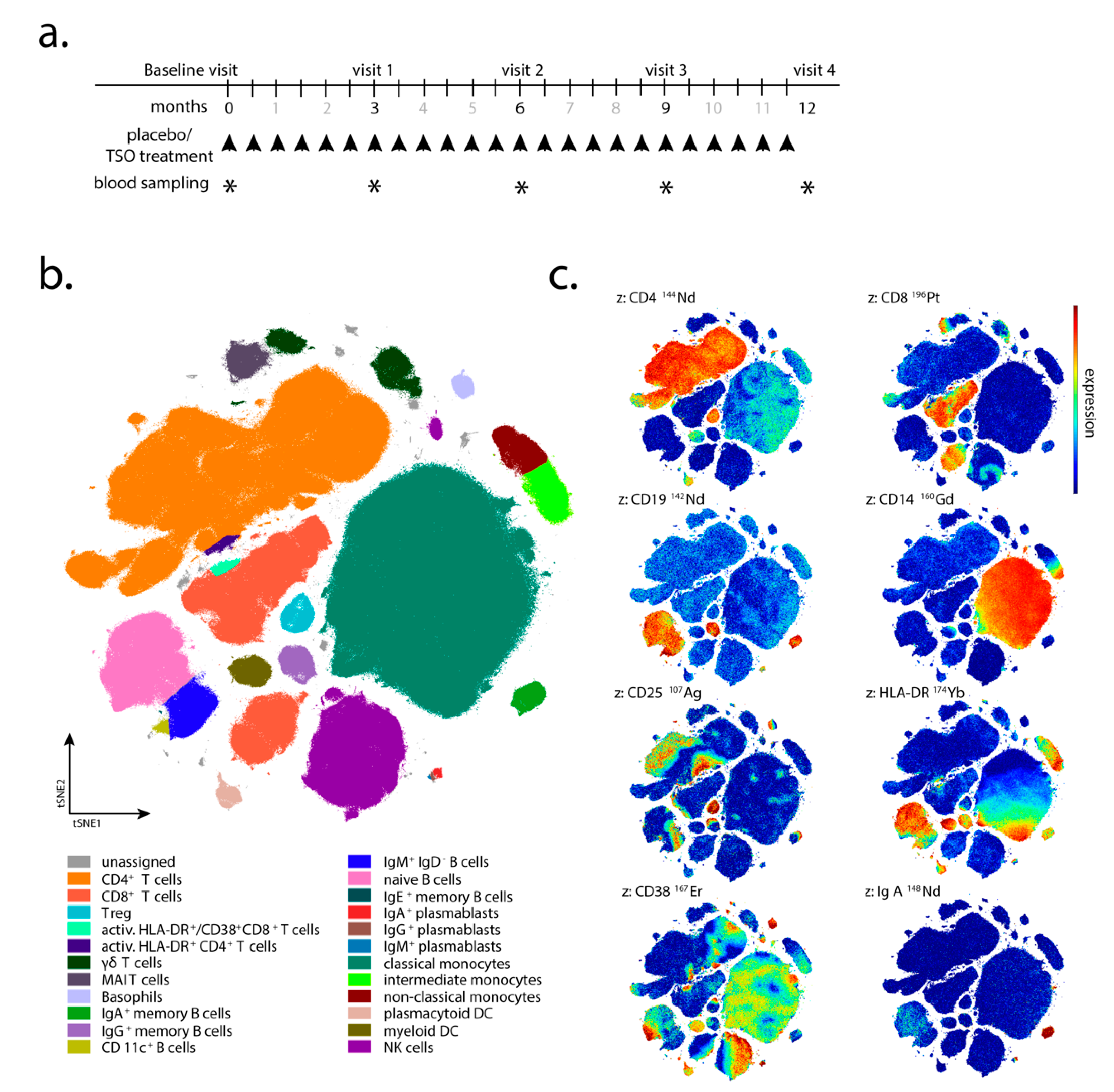
Trichuris suis ova (TSO) or placebo every 2 weeks for 12 months (
Figure 1a). The TSO suspension (sterile phosphate buffered saline, PBS) and placebo aliquots were supplied by OVAMED GmbH (Barsbüttel, Germany).
Patient visits were carried out after 3, 6, 9, and 12 months after the start of TRIOMS study (V1-V4;
Figure 1a). The primary clinical endpoint was the cumulative number of new hyperintense lesions identified on T2-weighted brain MRI during the treatment period of 12 months. Apart from treatment safety and tolerability, other secondary clinical endpoints included the annualized relapse rate and the proportion of relapse-free patients. In vivo mechanisms of action of TSO in the patients were assessed by extensive laboratory and immunological investigations. Due to low patient compliance with regular attendance of medical examinations and blood sampling, as well as limited sample/cell quantity collected at several sampling timepoints, not all immunological assays and clinical examinations could be performed for all patients and all timepoints of the study. Therefore, for serological analysis, immunoblotting, flow cytometry, and in vitro expansion and restimulation samples were available from four of five TSO-treated (002, 008, 010, 013) and four of six placebo-treated patients (003, 006, 011, 012) only. For mass cytometry, samples were available from four TSO-treated (002, 005, 008, 010) and six placebo-treated patients (001, 003, 004, 006, 011, 012). Due to limited patient sample size, we designed the investigation as a pilot study employing detailed immune-centered exploratory analysis at the individual patient level.
2.2. Peripheral Blood Mononuclear Cell (PBMC) Isolation
Briefly, 20–40 mL of peripheral blood were collected per visit in heparin tubes. Peripheral blood mononuclear cells (PBMC) were isolated via density gradient centrifugation over human Pancoll (1.077 g/mL, PAN-Biotech, Aidenbach, Germany). PBMC aliquots were cryopreserved at −80 °C in RPMI-1640 supplemented with 10% DMSO and 10% FCS until further use.
2.3. Mass Cytometry and Data Analysis
PBS and CyTOF staining medium (CSM) used in mass cytometric assays were prepared as described before [
19]. All antibodies and metal isotopes and their sources are listed in
Supplementary Table S1. In-house antibody conjugations with lanthanide and indium isotopes were carried out using MAXPAR X8 labeling kits (Fluidigm, Markham, ON, Canada) according to the manufacturer’s instructions. Platinum or palladium labeling was performed as described [
20,
21]. Cisplatins carrying isotopically enriched Pt were purchased from or kindly provided by Fluidigm. Cell events were detected using an iridium-containing DNA intercalator (Fluidigm). m-DOTA-Rh, used for discrimination of dead cells, was prepared from DOTA-maleimide (Macrocyclics, Dallas, TX, USA) and rhodium chloride (Sigma-Aldrich) as described [
22] and stored at 4 °C. Antibody–metal conjugates were used at concentrations optimized to separate cell populations expressing or lacking a particular antigen. All antibody cocktails were prepared in advance and cryopreserved at −80 °C [
19].
Cryopreserved PBMC were thawed as described [
19] and cell-surface-barcoded using a set of palladium- and platinum-isotope-labeled β2-microglobulin (B2M) antibodies [
20,
21]. Samples were run in a total of two pools of barcoded samples, prepared and acquired 1 week apart. Longitudinal samples of a given donor were always acquired as parts of the same sample pool. For this, PBMC were incubated for 30 min at 4 °C with combinations of B2M conjugates in a 5-choose-2 barcode scheme. Next, the barcoded cells were washed four times with 1.2 mL CSM (500×
g, 5 min, 4 °C), pooled and further processed together. Up to 17 × 10
6 pooled PBMC were stained in a 200 µL reaction with antibody cocktail 1 (Supplem.
Table 1) for 30 min at 4 °C and washed twice with 1 mL CSM (500×
g, 5 min, 4 °C). This was followed by a second 200 µL staining reaction with antibody cocktail 2 (Supplem.
Table 1) that included streptavidin-conjugated silver nanoparticles [
23]. After an incubation of 30 min at 4 °C, the PBMC pool was washed once with 1 mL PBS (500×
g, 5 min, 4 °C), and incubated in 1 mL 10 µM mDOTA-
103Rh solution in PBS for 5 min at room temperature (RT) to label dead cells for their later exclusion. The sample volume was filled up to 1500 µL with CSM followed by cell pelleting and supernatant aspiration. Then, the cells were washed once with 1 mL CSM, once with 1 mL PBS, and finally resuspended in a 2% PFA solution (diluted from 16% stock with PBS, Electron Microscopy Sciences, Hatfield, PA) and incubated at 4 °C overnight. On the next day, 500 µL CSM were added, cells were pelleted (700×
g, 5 min, 4 °C) and subsequently washed once with 1 mL CSM. Next, the sample pool was incubated for 25 min at room temperature (RT) in 500 µL 1 × permeabilization buffer (diluted with Millipore water from 10 × permeabilization buffer; Thermo Fisher Scientific, Waltham, MA, USA) supplemented 1:500
v/
v with 0.125 µM iridium-based DNA intercalator. Cells were then washed with 1 mL CSM. Thereafter, samples were resuspended in 1 mL PBS and counted with a MACSQuant flow cytometer (Miltenyi Biotec, Bergisch-Gladbach, Germany). Prior to acquisition on the CyTOF instrument (Fluidigm), samples were washed twice with 1 mL Millipore water and pelleted by centrifugation at 800×
g, 5 min, 4 °C. Cells were then resuspended in an appropriate volume of Millipore water to a maximum of 5 × 10
5 cells/mL supplemented with EQ 4 element calibration beads (1:10,
v/v) (Fluidigm) and filtered through 35 µm cell strainer cap tubes (BD, San Jose, CA, USA) prior to acquisition.
Mass cytometry was performed on CyTOF version 1 (operated as described before [
23,
24]) using instrument software version v6.0.626. The sample was acquired at a speed of 45 µL/min using the Supersampler injection system (Victorian Airship, Alamo, CA, USA). The instrument was run in dual calibration mode, with noise reduction turned on and event length thresholds set to 10 and 75. Raw data was converted to FCS 3.0 and normalized using EQ 4 element calibration beads and the CyTOF instrument software. Next, EQ 4 element calibration beads were excluded by gating on DNA(
193Ir)
+/
140Ce
– cell events in FlowJo (Version 10.4, TreeStar, Ashland, OR, USA). After further removal of cell aggregates and ion cloud fusion events according to DNA and cell length parameters, the pooled sample was deconvoluted using Boolean combinations of manual gates, as previously described [
25]. The resulting individual samples were further gated on CD45
+/Dead discriminator(
103Rh)
− cells and imported into the OMIQ.ai data analysis software (Santa Clara, CA, USA). An opt-SNE dimension reduction [
26] was performed with the markers indicated in
Supplementary Table S1 and manually drawn gates on the tSNE1/tSNE2 projection were applied. Statistical analysis was performed using GraphPad Prism Version 8 (San Diego, CA, USA).
2.4. Antibody Detection by Enzyme-Linked Immunosorbent Assay (ELISA)
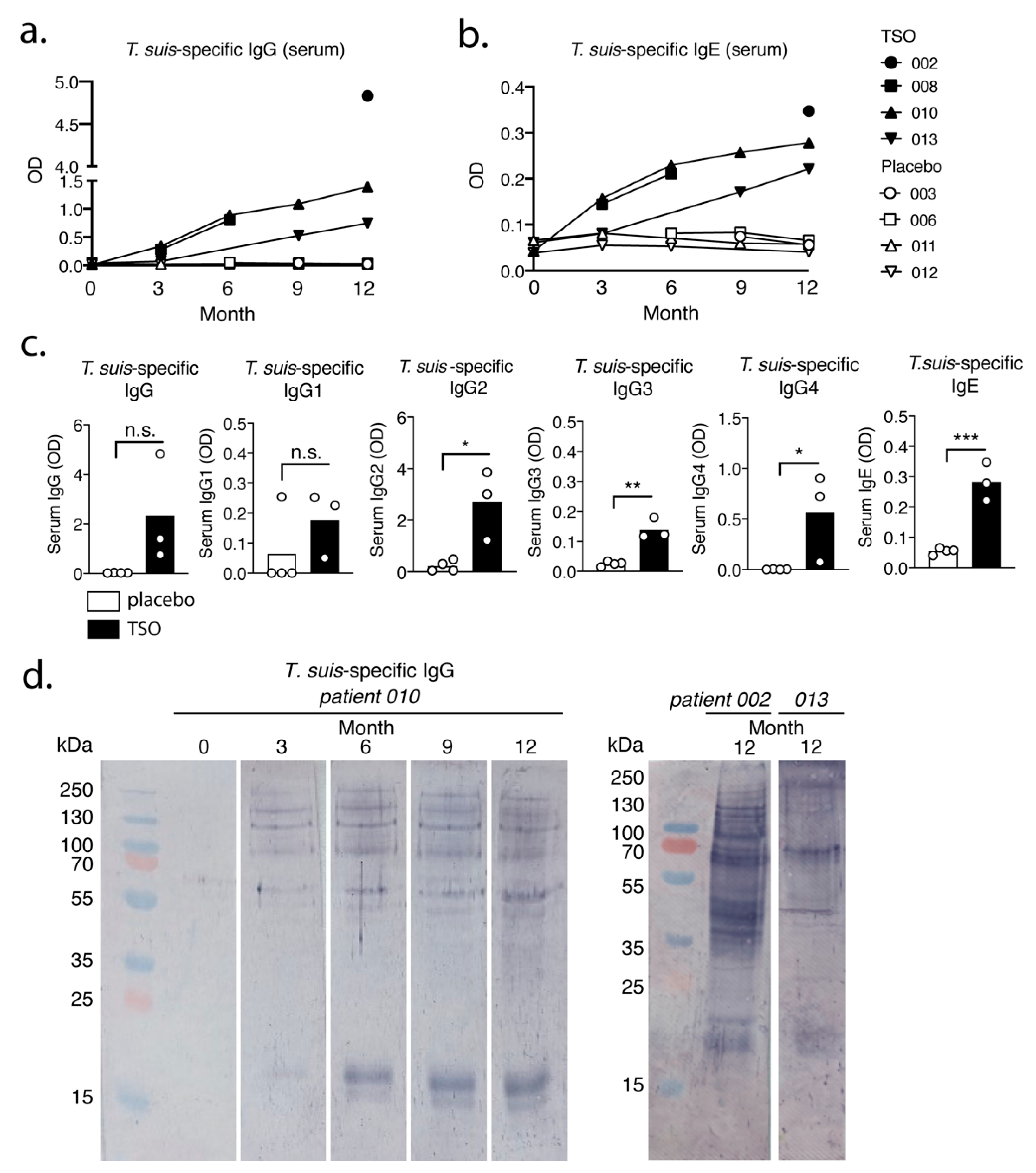
Total and T. suis-specific antibody titers in blood serum were quantified via sandwich ELISA. Briefly, 96-well flat-bottom Maxisorp plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated with 5 µg/mL T. suis adult excretory/secretory antigen (TsAdES) and were incubated overnight at 4 °C. Plates were washed using a Tecan Hydrospeed microplate washer and blocked with 200 µL 3% BSA diluted in PBS for 1 h at room temperature before 50 µL per well of samples were added. Internal standards for each assay were generated using a pooled serum sample from TSO-treated patients 002, 010, and 013 (Visit 4) as a reference positive control. Two-fold serial dilutions were performed on the reference positive control sample, with a top dilution of 1:2500 for IgG and 1:10 for IgG1-4 and IgE detection. Plates were incubated with the samples and the pooled reference control dilution series for 2 h at room temperature, after which 50 µL of the respective detection antibodies were added for 1 h. For IgE quantification, following sample incubation in the IgG4-coated plate, samples and blanks were transferred to the IgE-coated plate (IgG4 depletion step). The samples were then incubated in the IgE plate for 2 h at room temperature. After washing, 50 µL of POX substrate (1 tablet tetramethylbenzidine dihydrochloride in 10 mL phosphate-citrate buffer supplemented with 5 µL H2O2) or alkaline phosphatase (AP) substrate (1 tablet phosphatase substrate in 10 mL carbonate buffer supplemented with 10 µL 1 M MgCl2) were added for 15 min (POX) or 30 min (AP) and plates were incubated either for 15 min at room temperature (POX) or for 30 min at 37 °C (AP). To stop the enzymatic reaction, 25 µL of 1 M H2SO4 (POX) or 100 mM EDTA (AP) was added to each well and the signals was measured on a Biotek Synergy H1 Hybrid Reader. The following dilutions were used for serum samples: 1:10,000 for IgG and 1:20 for IgG1-4 and IgE. HRP-conjugated human IgG, IgG1, IgG3 were from OriGene (ME, USA), HRP-conjugated IgG4 from GeneTex (CA, USA), and AP-conjugated human IgE from Biomol (Hamburg, Germany). Parasite-specific antibody levels in serum are reported here as Optical Density (OD) values.
2.5. Generation of Parasite Antigen Material
Trichuris suis parasite antigens used to study Trichuris-specific antibody responses in serum by ELISA and Western blotting, as well as for the expansion and restimulation of Trichuris-specific T cells in vitro were generated as follows: T. suis adult worms were manually collected from the caecum and colon of T. suis-infected pigs. Worms were washed four times for 15 min each in RPMI-1640 (PAN-Biotech, Aidenbach, Germany) supplemented with 200 U/mL penicillin, 200 µg/mL Streptomycin, 1.35 µg/mL Amphotericin B (all purchased from PAN-Biotech) by sedimentation and were incubated overnight in RPMI-1640 supplemented with 1% Glucose (Sigma-Aldrich, St. Louis, US), 100 U/mL Penicillin, 100 µg/mL Streptomycin and 0.625 µg/mL Amphotericin B. Whole adult worm lysate (TsAd) antigen was produced by snap freezing the parasites and homogenization using a mortar and pestle. The homogenized material was then dissolved in PBS, filter sterilized and stored at −20°C until further use. For the generation of Trichuris excretory-secretory (antigens (TsAdES), culture medium was replaced and worms were incubated for another 5 days, while conditioned media was collected every 2nd day, filter-sterilized (0.45 µm Minisart Syringe Filter, Sartorius AG, Göttingen, Germany) and stored at −20 °C until further use. Filter-sterilized conditioned media from both male and female T. suis was further concentrated using centrifugal concentrators (Vivaspin, 5 KDa cut off, Sartorius). For the generation of L1 larval antigen (TsL1), freshly hatched larvae were hatched from embryonated TSO and were snap frozen and homogenized. The material was then dissolved in PBS and stored at -20°C until further use.
2.6. Immunoblot Analysis
SDS-PAGE protein separation was performed using a 12% separating gel and a 6% stacking gel. A total of 15 µg of TsAdES antigen was applied per lane. Following separation, proteins were transferred to a nitrocellulose membrane (1 h at 80 mA). Membranes were blocked for 1 h at room temperature with 5% non-fat dry milk in TBS/0.2% Tween buffer. Following the blocking step, membranes were washed three times in TBS/0.2% Tween (TBST) and were incubated overnight on a shaker at 4 °C with serum samples (diluted 1:200 in TBST supplemented with 5% dry milk). After overnight incubation, membranes were washed three more times in TBST buffer and were then incubated for 1 h at room temperature with AP-conjugated goat anti-human IgG (diluted 1:2000 in TBST/5% dry milk) (Sigma Aldrich), followed by washing three times in TBST buffer. For AP development, membranes were incubated for 5–10 min at RT in the dark with 10 mL AP development buffer, containing 66 µL p-nitroblautetrazoliumchloride (NBT) and 33 µL 5-brom-4-chlor-3-indolylphosphate (BCIP). The reaction was stopped by washing the membranes in distilled water.
2.7. Flow Cytometric Analysis
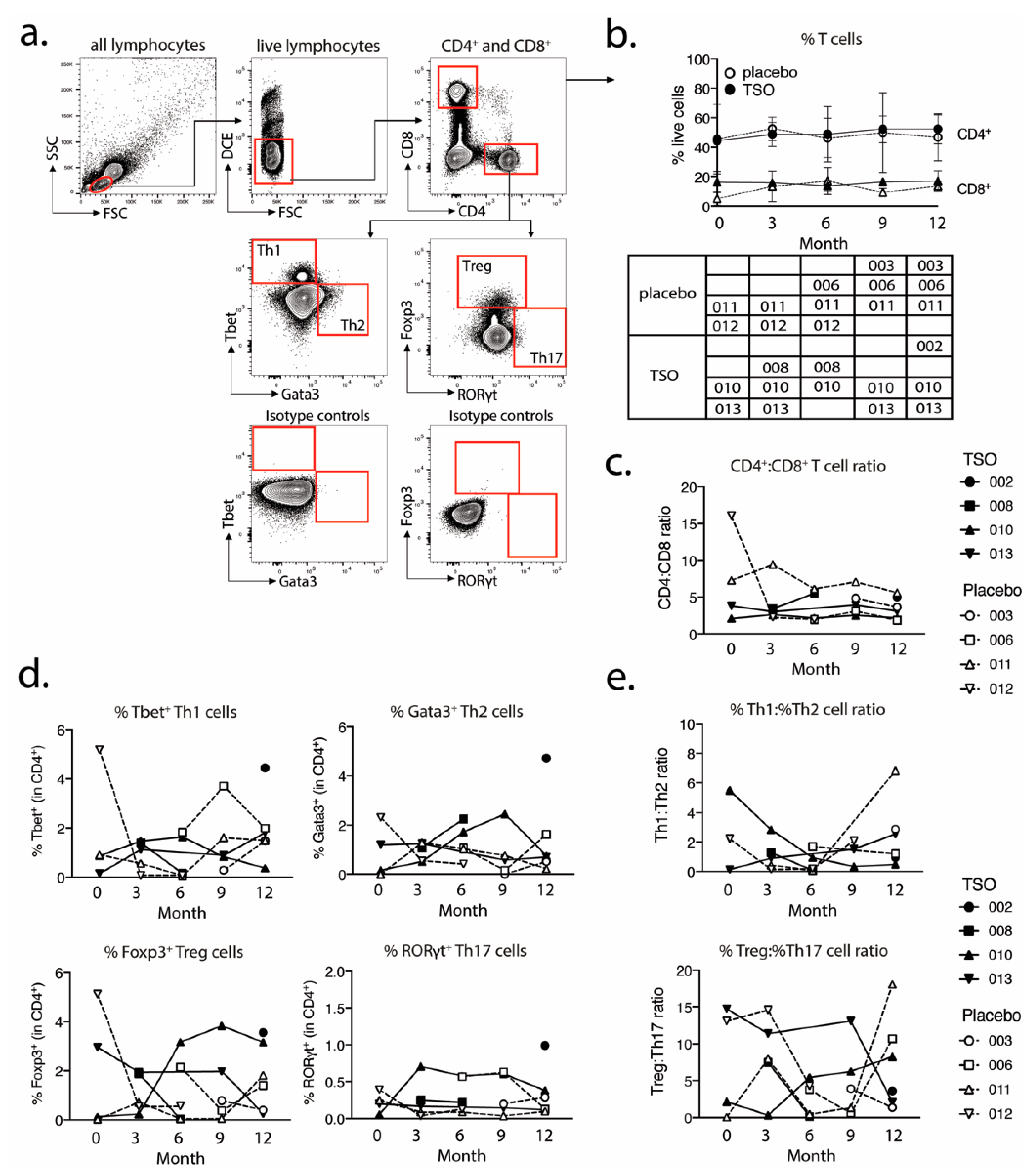
For the analysis of transcriptional factor expression in CD4+ and CD8+ T cells, 1 × 106 purified PBMCs were plated out per well in a 96-well conical bottom plate. For the intracellular staining of transcriptional factors, cells were fixed and permeabilized using the Fixation/Permeabilization buffer set (ThermoFisher/eBioscience). The following antibodies were used for the detection of cell surface and intracellular markers: CD4-PerCP-Cy5.5 (clone 0KT4), CD8-eF450 (clone OKT8), T-bet-PE-Cy7 (clone eBio4B10), GATA-3-eF660 (clone TWAJ), Foxp3-Alexa488 (clone 236A/E7), and RORγt-PE (clone AFKJS-9). In vitro expanded and restimulated PBMCs were stained using the same protocol and with the following markers for cell surface and intracellular marker detection: CD4-PerCP-Cy5.5 (clone 0KT4), CD8-eF450 (clone OKT8), IL-4-PE (clone 8D4-8), IL-10-PE (clone JES3-9D7), IL-13-APC (clone JES10-5A2), IL-17A-APC (clone eBio64DEC17), and IFNγ-eF660 (clone 4S.B3). Dead cells were excluded based on labeling with dead cell exclusion dye (Fixable Viability Dye eFluor780TM, ThermoFisher Scientific). Following intracellular staining, the cells were fixed in 200 µL 0.5% formaldehyde solution and were acquired on a BD Canto II (BD Biosciences). Antibodies were purchased from eBioscience (CD4, CD8, IL-17), ThermoFischer Scientific (IL-4, IFNγ, Fixable Viability Dye eFluor780TM), Biolegend (IL-10), and Miltenyi Biotec (IL-13).
2.8. In Vitro PBMC Expansion and Restimulation
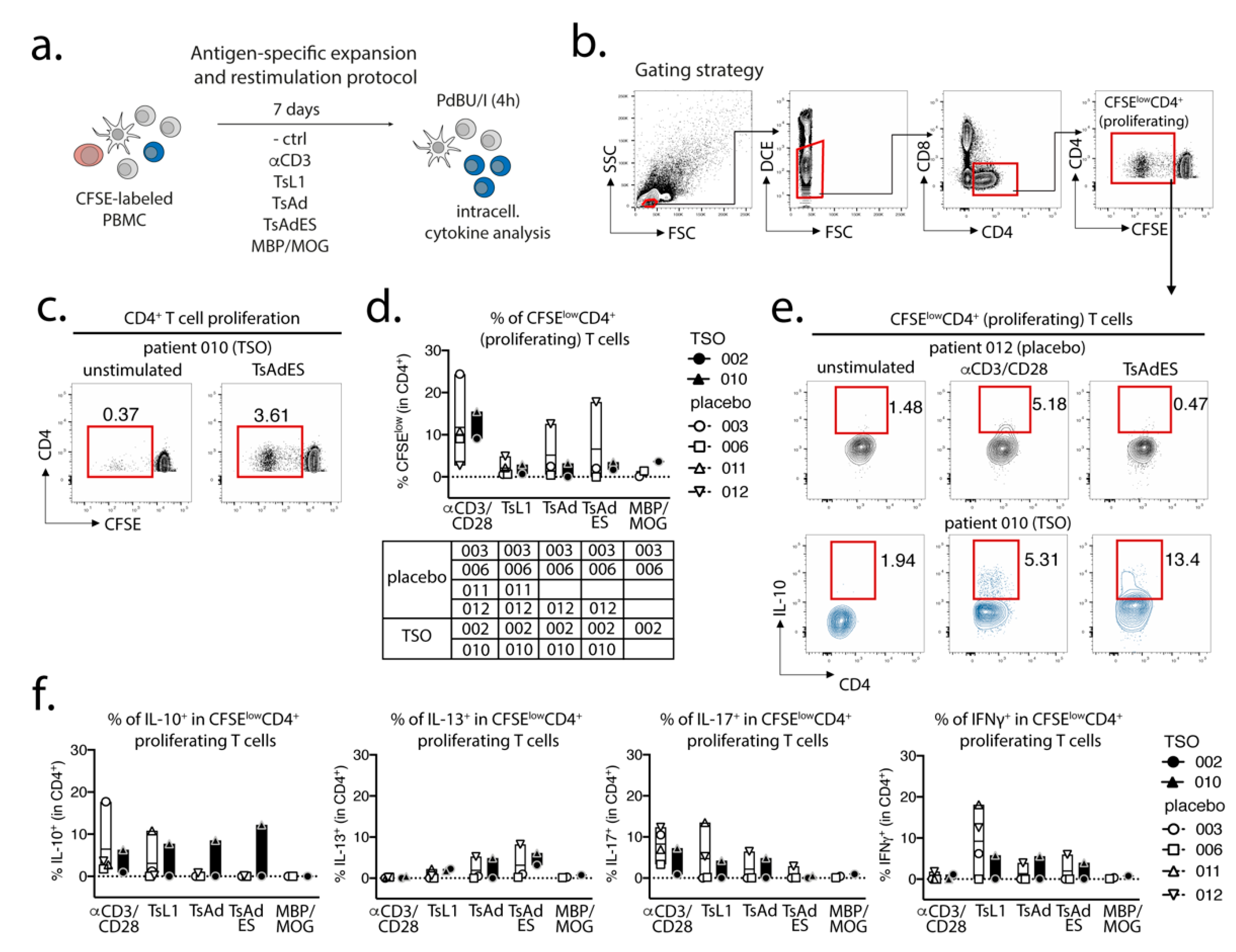
Prior to in vitro expansion and restimulation, single cell suspensions were prelabeled with CFSE (diluted 1:1000 in 1 mL PBS) for 8 min at RT in the dark and were washed in 20 mL AB HS buffer (RPMI 1640, 5% human serum, 1% sodium pyruvate, 1% nonessential amino acids, 5% penicillin/streptomycin, 5% L- glutamine). CFSE-labeled cells were then plated out in a 96-well round-bottom plate at a concentration of 1 × 106 cells per well in 200 µL complete medium (RPMI-1640, 5% human serum, 1% sodium pyruvate, 1% non-essential amino acids, 1% penicillin/streptomycin, 1% L-glutamine). PBMCs were expanded for 7 days at 37 °C, 5 % CO2 under the following conditions: unstimulated control (medium), αCD3 (2 µg/mL, clone Okt3), T. suis L1 stage larval antigen (TsL1, 5 µg/mL), T. suis crude adult antigen (TsAd, 20 µg/mL), T. suis adult excretory/secretory antigens (TsAdES, 20 µg/mL), MBP/MOG peptide mix (5 mM, Miltenyi Biotec). For restimulation on day 7, PBMCs were restimulated with PdBU (1:100,000) and Ionomycin (1:1000) for 30 min at 37 °C, 5% CO2, followed by another 3 h incubation with PdBU (1:100,000) and Brefeldin A (1:500) (ThermoFisher/Invitrogen).
2.9. Statistical Analysis
For assays and measurements, where an appropriate number of patient samples were available, statistical analysis was performed using GraphPad Prism Version 8 (San Diego, CA, USA). Results were tested for normal distribution using the Shapiro–Wilk normality tests, followed by an unpaired t-test or Mann–Whitney U test. Statistical significance is indicated as * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
Helminth-based treatment and more specifically, the use of
Trichuris suis eggs (TSO) to target various autoimmune diseases or inflammatory conditions have recently drawn considerable controversy. The inverse prevalence rates of chronic parasitic helminth infections and autoimmune diseases like multiple sclerosis (MS) across the world generally implies that the absence of naturally immunoregulatory active helminth infections in higher income communities and regions exacerbates the risk of autoimmune disease development and its clinical outcomes, including MS [
9,
27,
28,
29]. Nevertheless, robust clinical evidence of the efficacy of helminth treatment in interventional phase 2 studies is currently lacking [
13,
14].
MS is a multifaceted disease, comprising a range of disease subtypes [
30,
31], and with a variety of confounding factors involved in susceptibility, risk stratification, and treatment response capacities [
32,
33]. MS-centered studies therefore require strictly defined inclusion criteria, properly matched controls, and relatively large patient cohorts for clinical intervention studies. Although the TRIOMS study did not enroll a patient cohort large enough to fully evaluate treatment efficacy for the indicated clinical and radiographic endpoints, we employed a detailed exploratory immunological analysis at the individual patient level. Recently constructed genetic mapping of MS could identify the enrichment of MS susceptibility genes in various innate and adaptive immune cell populations, highlighting the potential contribution of functional shifts in one or more immune cell types to susceptibility to MS [
33]. In the context of experimental treatments such as helminth-based therapy, however, a detailed survey of interindividual differences in parasite-specific immune responses at the cellular level had not been performed to date. For that purpose, we specifically looked into the individual cellular and humoral adaptive immune responses against several
T. suis protein antigens. We combined global PBMC cell-surface marker expression-based immune phenotyping by mass cytometry with a more focused approach to address T helper cell subset differentiation, as well as parasite-specific antibody responses and T cell recall responses. A central objective was to delineate any stereotypical versus individual adaptive immune responses to TSO treatment in MS patients.
Our data suggest that the biweekly intake of 2500 TSO over 3–12 month consistently induces
T. suis antigen-specific antibodies of the subclasses IgG1-4 and IgE in all patients receiving TSO treatment. Importantly, this is in line with other studies demonstrating distinct and diverse antibody responses to natural infection with gastrointestinal nematodes [
34,
35], as well as with clinical studies applying experimental helminth infections as treatment [
13]. Moreover, for two TSO patients we observed a continuous elevation in
T. suis-specific IgG and IgE levels during the 12-month study and in one of those patients we could show a corresponding diversifying parasite-specific IgG repertoire over time via immunoblotting. For any heterogeneous population, including the TRIOMS patient cohort, some degree of variability in humoral immune responses to any nematode is expected. Likewise, Haswell-Elkins reported on considerable heterogeneity in the antibody recognition profiles of
Ascaris lumbricoides infected individuals in endemic regions, even when considering individuals from one household [
36]. Overall, we could therefore demonstrate that MS patients mount normal parasite-specific antibody responses against
T. suis as a result of prolonged TSO treatment over the course of 12 months.
Whether the continuous elevation of
T. suis specific antibody titers in two patients can be explained by the cumulative antigen availability due to repetitive treatment, or potentially further larval development and hence antigen variation, cannot be conclusively answered, but has been demonstrated also by others [
37]. It is likely that multiple rounds of exposure result in a continuous, but transient fluctuation of larval numbers in the patient’s gut and thus continuously increasing antibody titers might reflect increasing infection intensity over time as seen in natural
T. trichiura infections [
38]. Importantly,
T. suis typically presents as a self-limiting infection in humans, with no indications of adult worms or ova developing during TSO therapy [
39,
40]. The antibody recognition analysis of one patient (patient 010) suggests predominantly a quantitative development of the antibody repertoire.
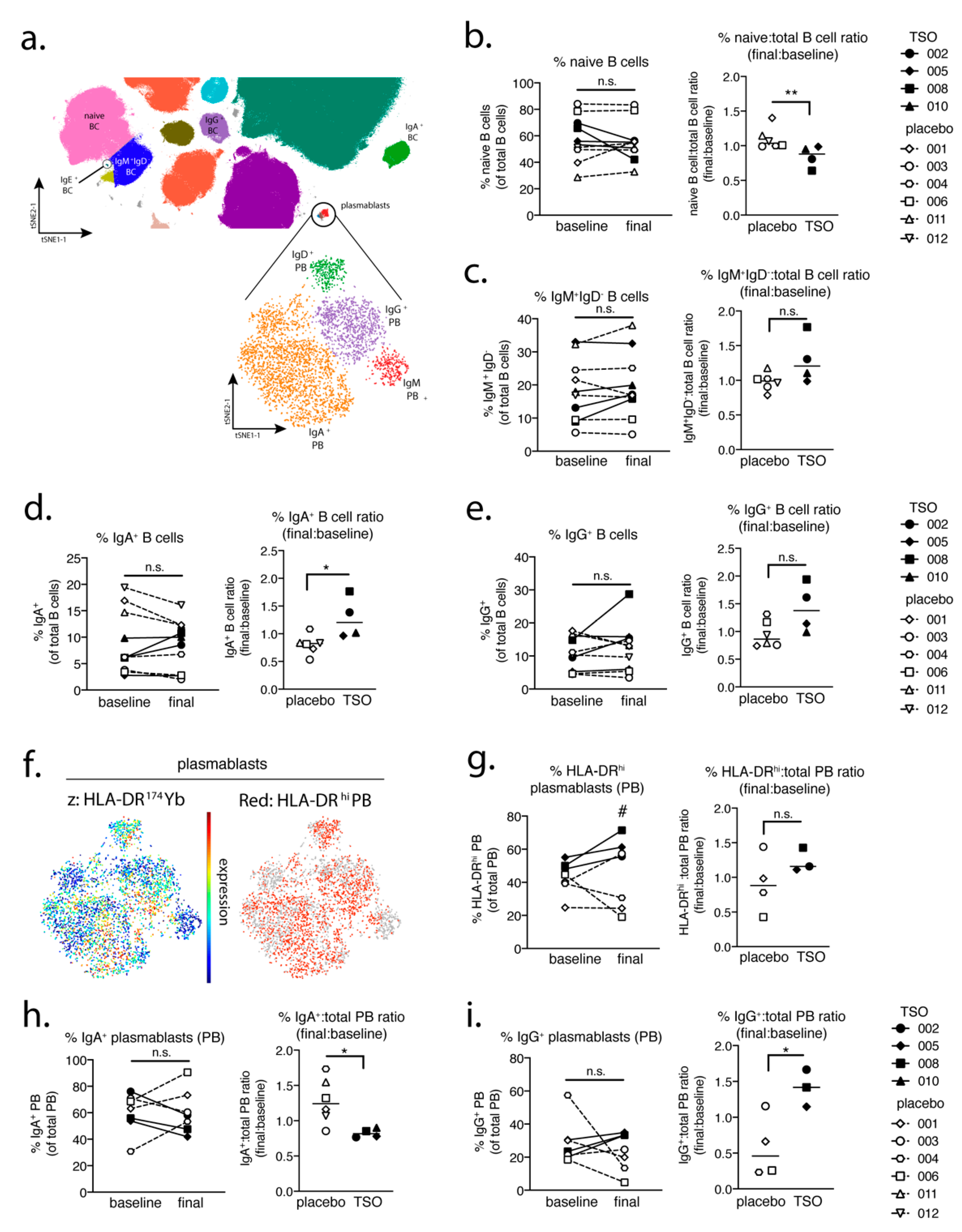
In a parallel approach, we employed a global PBMC immune phenotyping to study the cellular effects of TSO treatment on the basis of a baseline vs. post-treatment (final) comparison. Related to the increase of TSO-induced specific antibodies, our data show, on an individual level, the expansion of circulating IgG- and IgA-switched memory B cells, IgG+ plasmablasts, and HLA-DR+ plasmablasts, thus supporting TSO treatment effects on B cell responses. In turn, we observed no obvious baseline to final changes in the overall frequencies of natural killer (NK) cells, monocytes, basophils, dendritic cell (DC) subsets, γδ T cells, or mucosal-associated invariant T (MAIT) cells in either patient treatment group.
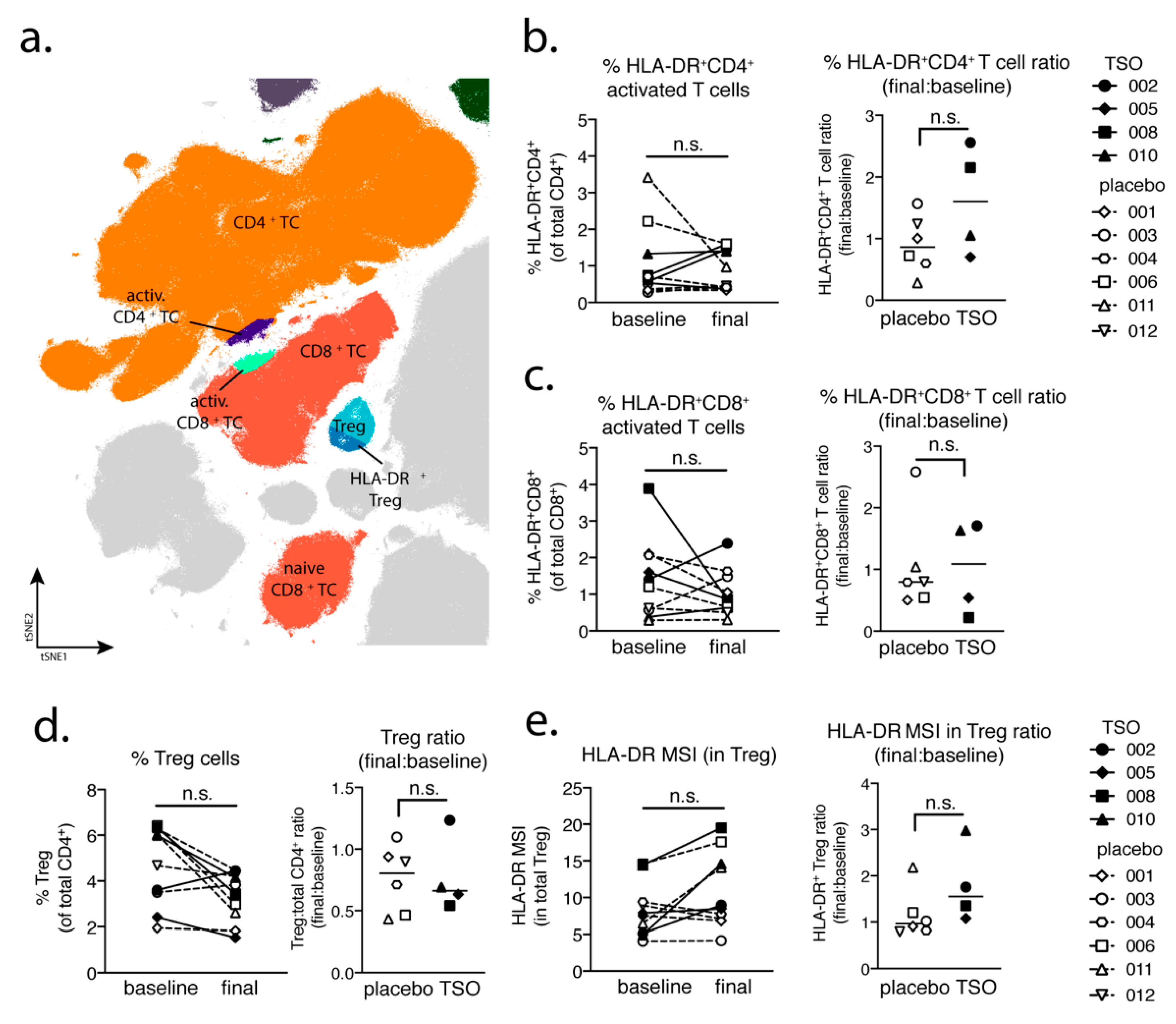
Studying the T cell arm in the adaptive immune response of MS patients receiving TSO therapy is obviously hampered by disease-associated hyperactivity and metabolic alterations of T cells that result in highly individual and fluctuating T cell activation signatures in context of disease progression [
41,
42]. This is reflected when looking at T cells from a global perspective using multiparameter phenotyping. The baseline vs. final timepoint comparison of activated CD8
+ cell frequencies varies greatly amongst individuals. However, our mass cytometry data revealed an increase of activated CD4
+ T cells in two out of four patients receiving TSO, an effect that is less pronounced in the group of placebo-treated MS patients. Nevertheless, due to the small sample size, more substantial conclusions could not be made on the basis of these findings.
We performed further intranuclear transcription factor analysis to dissect the TSO-induced CD4
+ T helper cell responses and their development over time. Again, we observed highly diverse kinetics of Tbet-expressing Th1 or GATA3
+ Th2 cells, but a relatively stable population of RORγt
+ Th17 cells within both the TSO- and the placebo-treated group. The induction of GATA3
+ Th2 cells is a prototypical host immune response to helminth infections and the Th1/Th2 ratio is supposed to be a crucial point in assessing the effects of helminth therapy on Th1-driven inflammatory conditions. A recent study has addressed the heterogeneity of human Th2 cells during helminth infection in more detail [
43]. They describe a decrease in a particular subset of CD27
−CD161
+ Th2 subset after deworming of helminth-infected individuals, which as a more pathogenic Th2 subset with enhanced effector functions potentially exert specific regulatory functions [
43]. Their results therefore indicate that, in order to dissect the potentially balancing effects of Th2 versus Th1 responses under TSO therapy, an even more detailed analysis of both subsets would be required.
Until now, the precise role of Treg cells in relapses and remission in MS is not completely understood and a functional impairment of specific Treg subsets is thought to be a major driver of relapses in MS patients [
44,
45,
46,
47]. On the other hand, in the context of helminth treatment, the induction of Treg is the most prominent pathway for parasite-driven immunomodulation (reviewed in [
48]). Our baseline vs. final comparison based on mass cytometry indicates no individual induction in Tregs for patients receiving TSO. Nevertheless, consistent with the overall CD4
+ T cell activation, we observed an increase in activated Treg cells, as evidenced by their elevated HLA-DR expression. Further analysis of CD39- and CD73-expressing Treg subsets did not reveal additional hints for Treg subset regulation by TSO (data not shown). In line with the dynamics of Th1 and Th2 subsets during the study, transcription factor analysis of Foxp3
+ CD4 T cells varied greatly over time in both patient groups, potentially reflecting the different clinical relapse courses of RRMS patients enrolled here.
In summary, the data presented here as part of a phase II pilot study of a limited cohort of MS patients receiving TSO as an alternative treatment approach (TRIOMS), indicates a high degree of interindividual variability in the adaptive immune responses to this helminth treatment. In light of the development of biologicals like TSO for the treatment of immune-mediated diseases like MS, future larger scale studies involving diverse MS patient cohorts should aim to better address and incorporate this interindividual variability of adaptive immune responses at the cellular level into any personalized medicine framework. For future studies focusing on the further development and optimization of helminth-based treatment for autoimmune diseases, the focus should therefore include a tailored treatment in order to meet the highly variable individual host cellular responses and to identify treatment “responder” and “nonresponder” patients, in particular in multifaceted diseases like MS. Biologicals like TSO might be highly beneficial for subgroups of MS patients, but to achieve this goal precision medicine probably is the choice to improve clinical treatment efficacy.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}