Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene–Implication for Cardiovascular Health

Abstract
1. Introduction
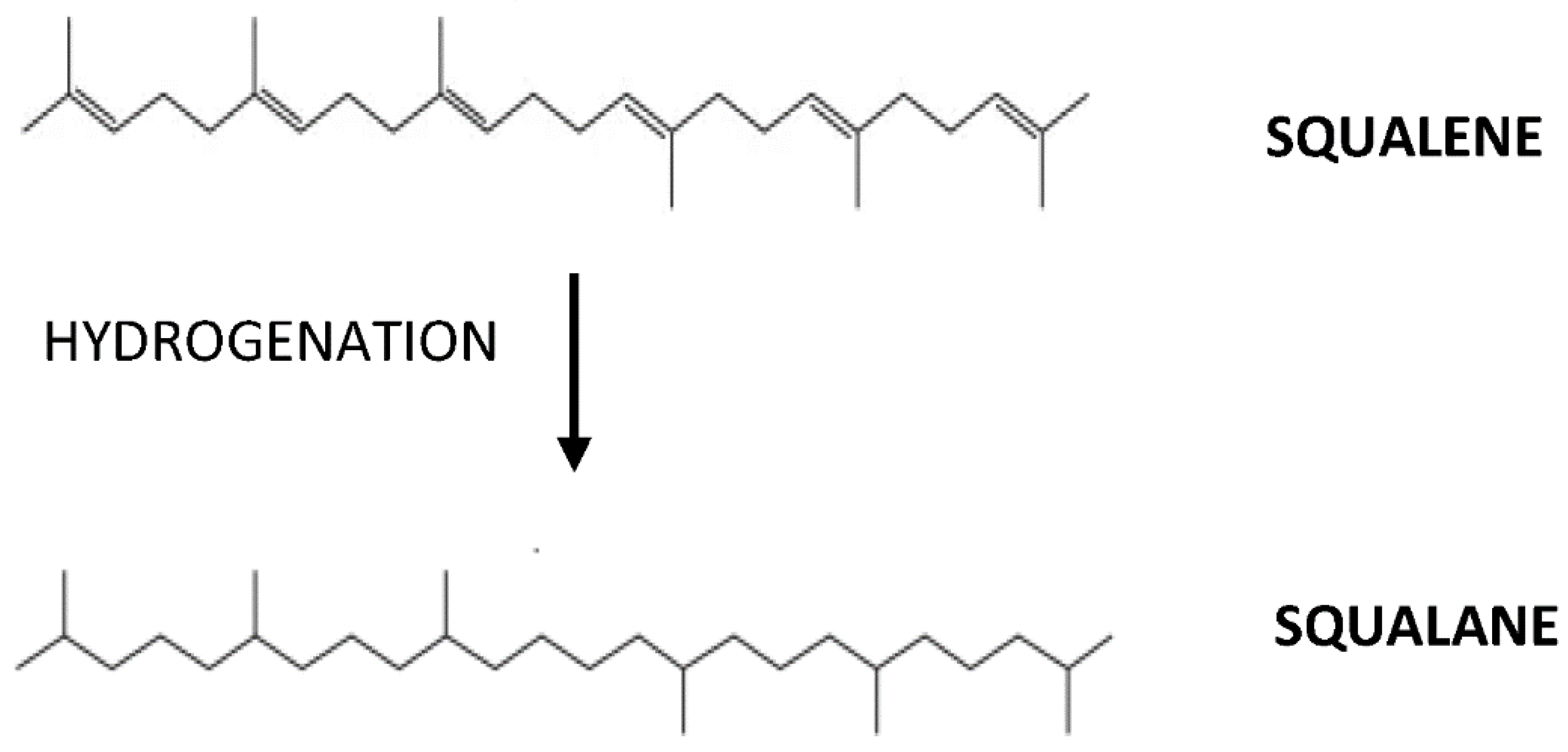
2. Antioxidant Activity of Squalene Related to Cardiovascular Health
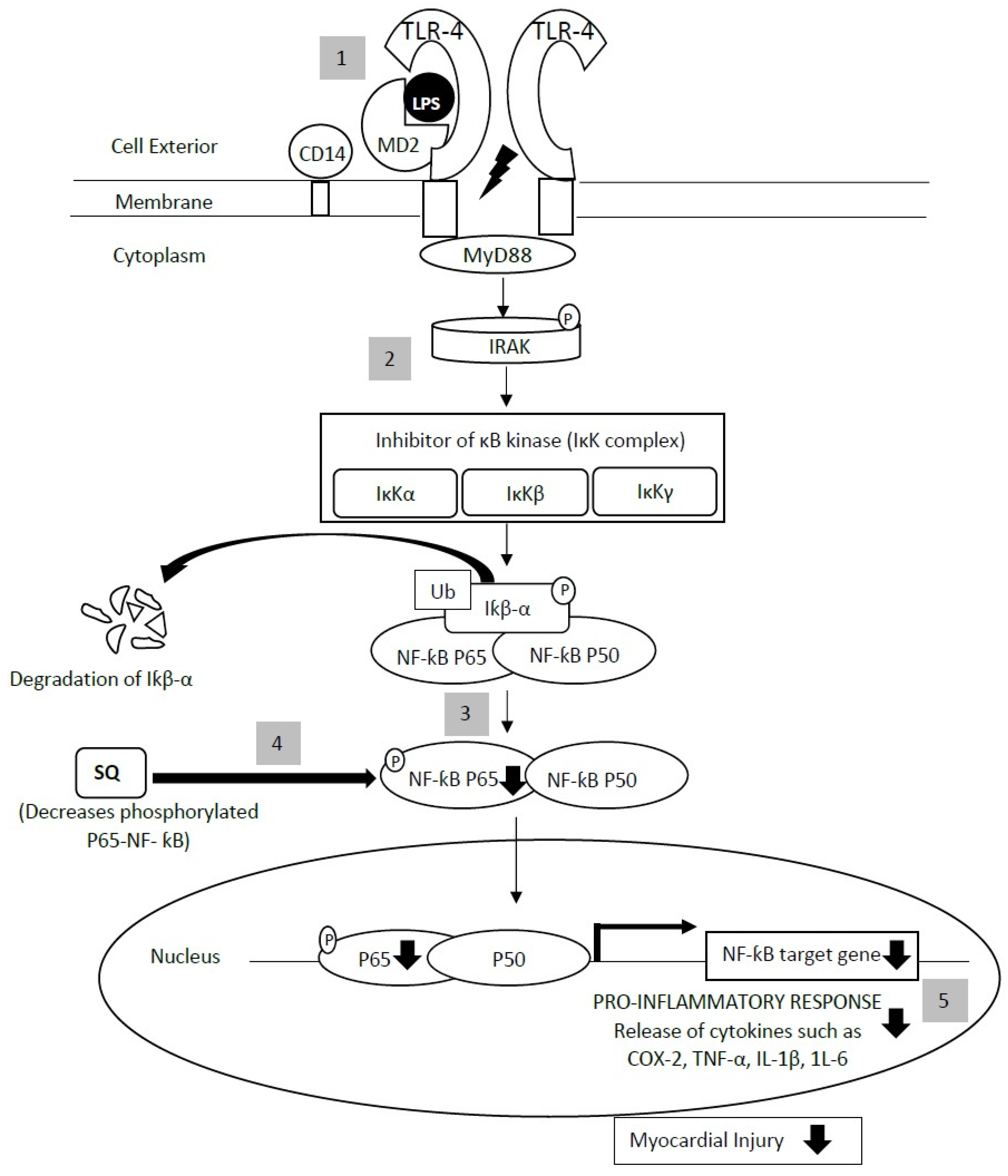
3. Anti-Inflammatory Actions of Squalene
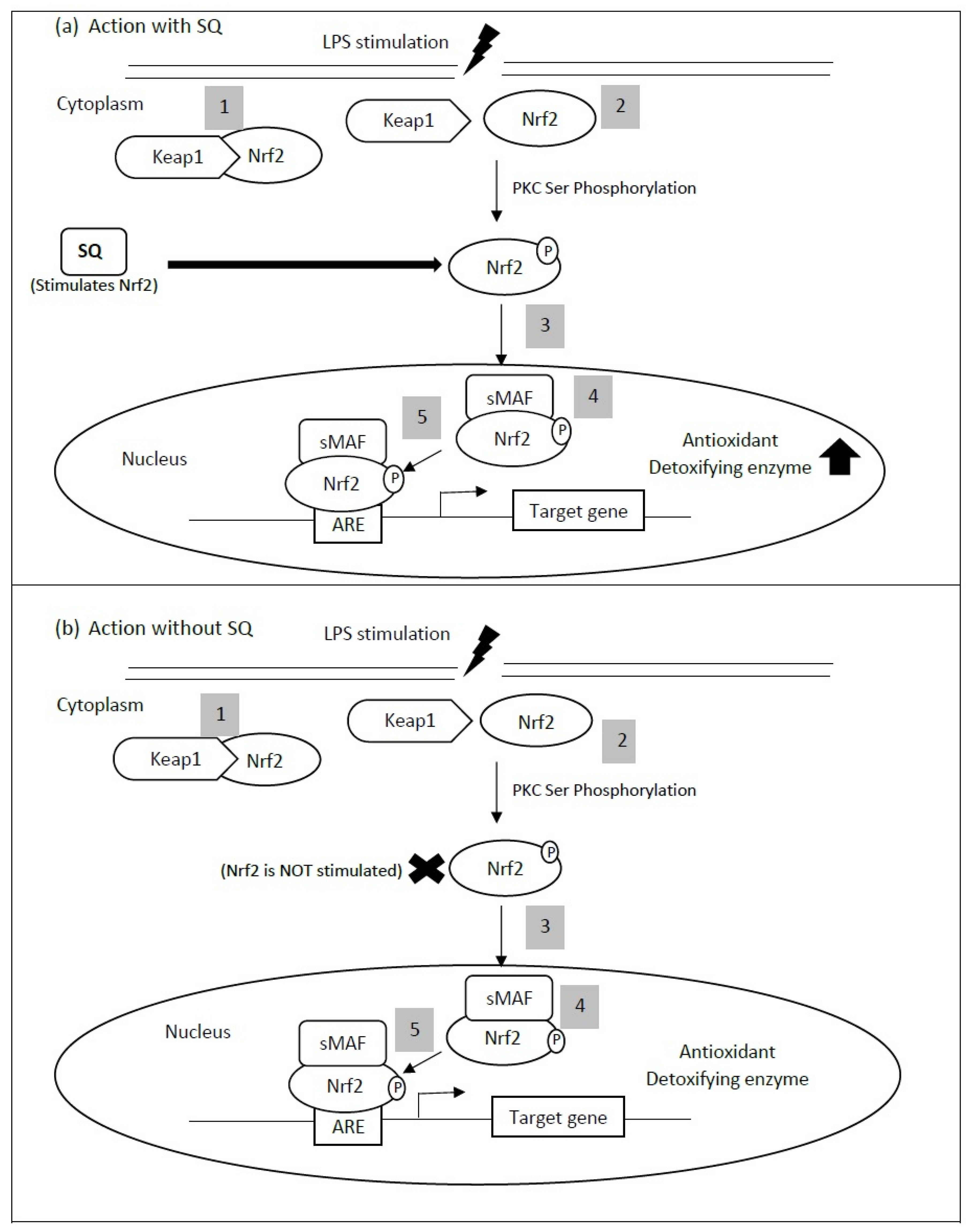
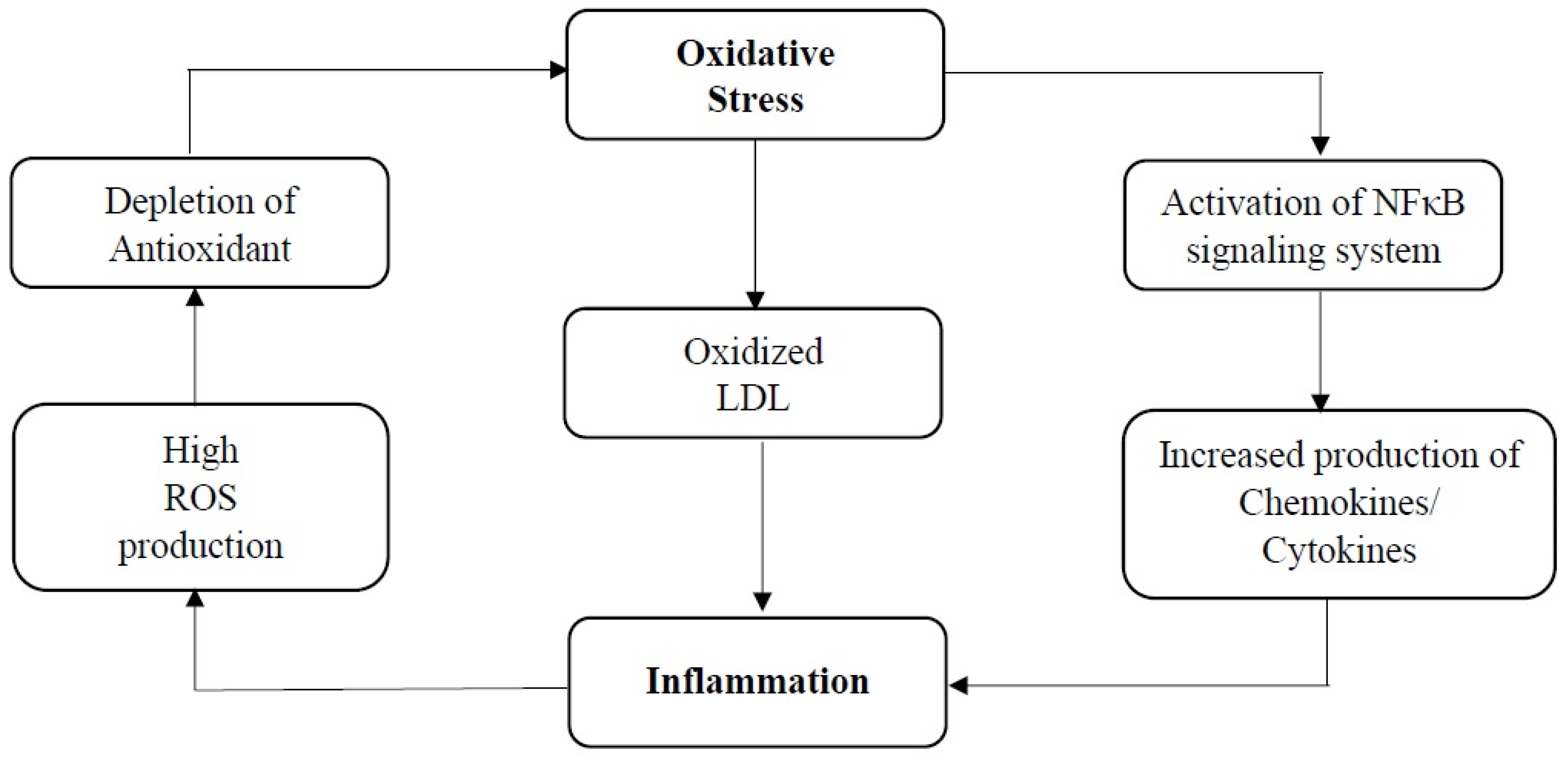
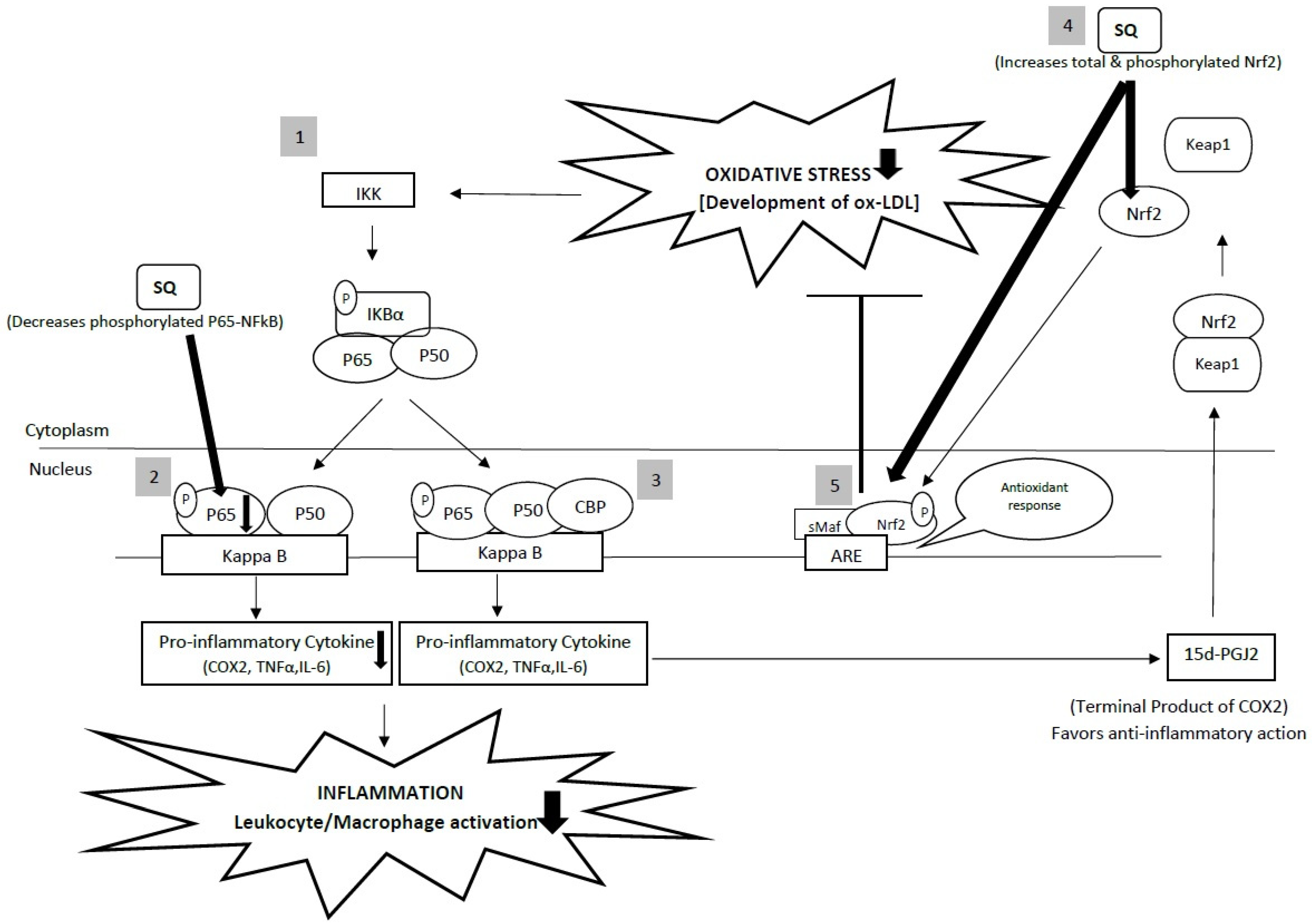
4. Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene in Cardiovascular Health
5. Possible Application of Squalene Supplementation in Humans
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Cardiovascular Disease. Available online: https://www.who.int/health-topics/cardiovascular-diseases/ (accessed on 5 November 2020).
- Stewart, J.; Manmathan, G.; Wilkinson, P. Primary prevention of cardiovascular disease: A review of contemporary guidance and literature. JRSM Cardiovasc. Dis. 2017, 6, 2048004016687211. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Challenge of Cardiovascular Disease—Quick Statistics. Available online: http://www.euro.who.int/en/health-topics/noncommunicable-diseases/cardiovascular-diseases/data-and-statistics (accessed on 16 November 2020).
- Schenck-Gustafsson, K. Traditional Cardiovascular Disease Risk Factors. In ESC CardioMed (3 edn); Camm, J.A., Lüscher, T.F., Maurer, G., Serruys, P.W., Eds.; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Yusuf, S.; Hawken, S.; Ôunpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Sudhakaran, S.; Bottiglieri, T.; Tecson, K.M.; Kluger, A.Y.; McCullough, R.O.P.A. Alteration of lipid metabolism in chronic kidney disease, the role of novel antihyperlipidemic agents, and future directions. Rev. Cardiovasc. Med. 2018, 19, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.H. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef]
- Yao, Y.S.; Di Li, T.; Zeng, Z.H. Mechanisms underlying direct actions of hyperlipidemia on myocardium: An updated review. Lipids Health Dis. 2020, 19, 23. [Google Scholar] [CrossRef]
- Zárate, A.; Manuel-Apolinar, L.; Saucedo, R.; Hernández-Valencia, M.; Basurto, L. Hypercholesterolemia as a Risk Factor for Cardiovascular Disease: Current Controversial Therapeutic Management. Arch. Med. Res. 2016, 47, 491–495. [Google Scholar] [CrossRef]
- Cheng, V.Y.; Berman, D.S.; Rozanski, A.; Dunning, A.M.; Achenbach, S.; Al-Mallah, M.; Budoff, M.J.; Cademartiri, F.; Callister, T.Q.; Chang, H.-J.; et al. Performance of the traditional age, sex, and angina typicality-based approach for estimating pretest probability of angiographically significant coronary artery disease in patients undergoing coronary computed tomographic angiography: Results from the multinational coronary CT angiography evaluation for clinical outcomes: An international multicenter registry (CONFIRM). Circulation 2011, 124, 2423–2432. [Google Scholar]
- Makris, A.; Foster, G.D. Dietary Approaches to the Treatment of Obesity. Psychiatr. Clin. N. Am. 2011, 34, 813–827. [Google Scholar] [CrossRef]
- Zodda, D.; Giammona, R.; Schifilliti, S. Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy 2018, 6, 10. [Google Scholar] [CrossRef]
- Last, A.R.; Ference, J.D.; Menzel, E.R. Hyperlipidemia: Drugs for Cardiovascular Risk Reduction in Adults. Am. Fam. Physician 2017, 95, 78–87. [Google Scholar]
- Safeer, R.S.; LaCivita, C.L. Choosing drug therapy for patients with hyperlipidemia. Am. Fam. Physician 2000, 61, 3371–3382. [Google Scholar] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Iii, W.H.S.; Khan, B.V.; Sperling, L.S. Management of the statin-intolerant patient. Curr. Treat. Options Cardiovasc. Med. 2009, 11, 263–271. [Google Scholar]
- Bouknight, P.; Mackler, L.; Heffington, M. FPIN’s clinical inquiries. Best alternatives to statins for treating hyperlipidemia. Am. Fam. Physician 2007, 76, 1027–1029. [Google Scholar]
- Wider, B.; Pittler, M.H.; Thompson-Coon, J.; Ernst, E. Artichoke leaf extract for treating hypercholesterolaemia. Cochrane Database Syst. Rev. 2013, 2013, CD003335. [Google Scholar] [CrossRef]
- Nies, L.K.; Cymbala, A.A.; Kasten, S.L.; Lamprecht, D.G.; Olson, K.L. Complementary and Alternative Therapies for the Management of Dyslipidemia. Ann. Pharmacother. 2006, 40, 1984–1992. [Google Scholar] [CrossRef]
- Ghimire, G.P.; Thuan, N.H.; Koirala, N.; Sohng, J.K. Advances in Biochemistry and Microbial Production of Squalene and Its Derivatives. J. Microbiol. Biotechnol. 2016, 26, 441–451. [Google Scholar] [CrossRef]
- Rohmer, M.; Seemann, M.; Horbach, S.; Bringer-Meyer, S.; Sahm, H. Glyceraldehyde 3-Phosphate and Pyruvate as Precursors of Isoprenic Units in an Alternative Non-Mevalonate Pathway for Terpenoid Biosynthesis. J. Am. Chem. Soc. 1996, 118, 2564–2566. [Google Scholar] [CrossRef]
- Wołosik, K.; Knaś, M.; Zalewska, A.; Niczyporuk, M.; Przystupa, A.W. The importance and perspective of plant-based squalene in cosmetology. J. Cosmet. Sci. 2013, 64, 59–66. [Google Scholar]
- Nowicki, R.; Barańska-Rybak, W. Shark liver oil as a supporting therapy in atopic dermatitis. Polski Merkur. Lek. 2007, 22, 312–313. [Google Scholar]
- Okada, K.; Matsumoto, K. Effect of Skin Care with an Emollient Containing a High Water Content on Mild Uremic Pruritus. Ther. Apher. Dial. 2004, 8, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Turchini, G.M.; Ng, W.-K.; Tocher, D.R. Fish Oil Replacement and Alternative Lipid Sources in Aquaculture Feeds; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Lozano-Grande, M.A.; Gorinstein, S.; Espitia-Rangel, E.; Dávila-Ortiz, G.; Martínez-Ayala, A.L. Plant Sources, Extraction Methods, and Uses of Squalene. Int. J. Agron. 2018, 2018, 1829160. [Google Scholar] [CrossRef]
- Pappas, A. Epidermal surface lipids. Dermato-Endocrinol. 2009, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Gunes, F. Medical use of squalene as a natural antioxidant. J. Marmara Univ. Inst. Health Sci. 2013, 3, 221–229. [Google Scholar]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Chan, P.; Tomlinson, B.; Lee, C.B.; Lee, Y.S. Effectiveness and safety of low-dose pravastatin and squalene, alone and in combination, in elderly patients with hypercholesterolemia. J. Clin. Pharmacol. 1996, 36, 422–427. [Google Scholar] [CrossRef]
- Ibrahim, N.; Fairus, S.; Zulfarina, M.S.; Mohamed, I.N. The Efficacy of Squalene in Cardiovascular Disease Risk-A Systematic Review. Nutrient 2020, 12, 414. [Google Scholar] [CrossRef]
- Kelly, G.S. Squalene and its potential clinical uses. Altern. Med. Rev. 1999, 4, 29–36. [Google Scholar]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Lou-Bonafonte, J.M.; Martínez-Beamonte, R.; Sanclemente, T.; Surra, J.C.; Herrera-Marcos, L.V.; Sanchez-Marco, J.; Arnal, C.; Osada, J. Current Insights into the Biological Action of Squalene. Mol. Nutr. Food Res. 2018, 62, e1800136. [Google Scholar] [CrossRef]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ou, B.; Prior, R.L. The Chemistry behind Antioxidant Capacity Assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Moharram, H.; Youssef, M. Methods for Determining the Antioxidant Activity: A Review. Alex. J. Food Sci. Technol. 2014, 11, 31–42. [Google Scholar]
- Gabás-Rivera, C.; Barranquero, C.; Martínez-Beamonte, R.; Navarro, M.A.; Surra, J.C.; Osada, J. Dietary Squalene Increases High Density Lipoprotein-Cholesterol and Paraoxonase 1 and Decreases Oxidative Stress in Mice. PLoS ONE 2014, 9, e104224. [Google Scholar] [CrossRef] [PubMed]
- Guillén, N.; Acín, S.; Navarro, M.A.; Perona, J.S.; Arbonés-Mainar, J.M.; Arnal, C.; Sarría, A.J.; Surra, J.C.; Carnicer, R.; Orman, I.; et al. Squalene in a sex-dependent manner modulates atherosclerotic lesion which correlates with hepatic fat content in apoE-knockout male mice. Atherosclerosis 2008, 197, 72–83. [Google Scholar] [CrossRef]
- Dhandapani, N.; Ganesan, B.; Anandan, R.; Jeyakumar, R.; Rajaprabhu, D.; Ezhilan, R.A. Synergistic effects of squalene and polyunsaturated fatty acid concentrate on lipid peroxidation and antioxidant status in isoprenaline-induced myocardial infarction in rats. Afr. J. Biotechnol. 2007, 6, 6. [Google Scholar]
- Farvin, K.S.; Anandan, R.; Kumar, S.H.S.; Shiny, K.; Mathew, S.; Sankar, T.; Nair, P.V. Cardioprotective Effect of Squalene on Lipid Profile in Isoprenaline-Induced Myocardial Infarction in Rats. J. Med. Food 2006, 9, 531–536. [Google Scholar] [CrossRef]
- Farvin, K.S.; Kumar, S.H.S.; Anandan, R.; Mathew, S.; Sankar, T.; Nair, P.V. Supplementation of squalene attenuates experimentally induced myocardial infarction in rats. Food Chem. 2007, 105, 1390–1395. [Google Scholar] [CrossRef]
- Farvin, K.H.S.; Anandan, R.; Sankar, T.V.; Nair, P.G.V. Protective Effect of Squalene against Isoproterenol-Induced Myocardial Infarction in Rats. J. Clin. Biochem. Nutr. 2005, 37, 55–60. [Google Scholar] [CrossRef]
- Farvin, K.S.; Anandan, R.; Kumar, S.H.S.; Shiny, K.S.; Sankar, T.V.; Thankappan, T.K. Effect of squalene on tissue defence system in isoproterenol-induced myocardial infarction in rats. Pharmacol. Res. 2004, 50, 231–236. [Google Scholar]
- Motawi, T.M.; Sadik, N.A.E.-H.; Refaat, A. Cytoprotective effects of DL-alpha-lipoic acid or squalene on cyclophosphamide-induced oxidative injury: An experimental study on rat myocardium, testicles and urinary bladder. Food Chem. Toxicol. 2010, 48, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Gordon, E.M.; Figueroa, D.M.; Barochia, A.V.; Levine, S.J. Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 55, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Boskou, D.; Clodoveo, M. Squalene: A Trove of Metabolic Actions. In Products from Olive Tree; Clodoveo, M., Boskou, D., Eds.; IntechOpen: London, UK, 2016. [Google Scholar]
- Mehdi, M.M.; Rizvi, S.I. Human Plasma Paraoxonase 1 (PON1) Arylesterase Activity During Aging: Correlation with Susceptibility of LDL Oxidation. Arch. Med. Res. 2012, 43, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Lee, J.-W.; Youn, Y.-J.; Ahn, M.-S.; Ahn, S.G.; Yoo, B.-S.; Lee, S.-H.; Yoon, J.; Choe, K.-H. Urinary Levels of 8-Iso-Prostaglandin F2α and 8-Hydroxydeoxyguanine as Markers of Oxidative Stress in Patients with Coronary Artery Disease. Korean Circ. J. 2012, 42, 614–617. [Google Scholar] [CrossRef][Green Version]
- Khoubnasabjafari, M.; Ansarin, K.; Jouyban, A. Reliability of malondialdehyde as a biomarker of oxidative stress in psychological disorders. BioImpacts 2015, 5, 123–127. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Górnicka, M.; Ciecierska, A.; Hamulka, J.; Drywień, M.E.; Frackiewicz, J.; Górnicki, K.; Wawrzyniak, A. α-Tocopherol Protects the Heart, Muscles, and Testes from Lipid Peroxidation in Growing Male Rats Subjected to Physical Efforts. Oxidative Med. Cell. Longev. 2019, 2019, 8431057. [Google Scholar] [CrossRef]
- Sishi, B.J.N. Chapter 10—Autophagy Upregulation Reduces Doxorubicin-Induced Cardiotoxicity, in Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 157–173. [Google Scholar]
- Chang, H.M.; Moudgil, R.; Scarabelli, T.; Okwuosa, T.M.; Yeh, E.T. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J. Am. Coll. Cardiol. 2017, 70, 2536–2551. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants 2019, 9, 4. [Google Scholar] [CrossRef]
- Cárdeno, A.; Aparicio-Soto, M.; La Paz, S.M.-D.; Bermudez, B.; Muriana, F.J.G.; Alarcón-De-La-Lastra, C. Squalene targets pro- and anti-inflammatory mediators and pathways to modulate over-activation of neutrophils, monocytes and macrophages. J. Funct. Foods 2015, 14, 779–790. [Google Scholar] [CrossRef]
- Vomhof-DeKrey, E.E.; Picklo, M.J. The Nrf2-antioxidant response element pathway: A target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent Modification at Cys151 Dissociates the Electrophile Sensor Keap1 from the Ubiquitin Ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.-J.; Kundu, J.K.; Na, H.-K. Nrf2 as a Master Redox Switch in Turning on the Cellular Signaling Involved in the Induction of Cytoprotective Genes by Some Chemopreventive Phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. Reexamination of the electrophile response element sequences and context reveals a lack of consensus in gene function. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2010, 1799, 496–501. [Google Scholar] [CrossRef][Green Version]
- Eggler, A.L.; Gay, K.A.; Mesecar, A.D. Molecular mechanisms of natural products in chemoprevention: Induction of cytoprotective enzymes by Nrf2. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S84–S94. [Google Scholar] [CrossRef]
- Howden, R. Nrf2 and Cardiovascular Defense. Oxidative Med. Cell. Longev. 2013, 2013, 104308. [Google Scholar] [CrossRef]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3’-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef]
- Wiesel, P.; Patel, A.P.; Carvajal, I.M.; Wang, Z.Y.; Pellacani, A.; Maemura, K.; Difonzo, N.; Rennke, H.G.; Layne, M.D.; Yet, S.-F.; et al. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ. Res. 2001, 88, 1088–1094. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Zhu, G.; Zhang, P.; Van Deel, E.; French, J.; Fassett, J.; Oury, T.; Bache, R.; et al. Extracellular Superoxide Dismutase Deficiency Exacerbates Pressure Overload–Induced Left Ventricular Hypertrophy and Dysfunction. Hypertension 2008, 51, 19–25. [Google Scholar] [CrossRef]
- Matsushima, S.; Kinugawa, S.; Ide, T.; Matsusaka, H.; Inoue, N.; Ohta, Y.; Yokota, T.; Sunagawa, K.; Tsutsui, H. Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2237–H2245. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 Protects against Maladaptive Cardiac Responses to Hemodynamic Stress. Arter. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Falk, E.; Palacios, I.F.; Newell, J.B.; Fuster, V.; Fallon, J.T. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation 1994, 90, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Willerson, J.T. Prospects for cardiovascular research. JAMA 2001, 285, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Simon, D.I. Inflammation and Thrombosis. Circulation 2001, 103, 1718–1720. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.; Vreeburg, R.A.M.; Savelkoul, H.F.J.; Wichers, H.J. Transcription profiles of LPS-stimulated THP-1 monocytes and macrophages: A tool to study inflammation modulating effects of food-derived compounds. Food Funct. 2010, 1, 254–261. [Google Scholar] [CrossRef]
- Liu, G.; Park, Y.J.; Abraham, E. Interleukin-1 receptor-associated kinase (IRAK) -1-mediated NF-kappaB activation requires cytosolic and nuclear activity. Faseb J. 2008, 22, 2285–2296. [Google Scholar] [CrossRef]
- Jung, K.H.; Hong, S.W.; Zheng, H.M.; Lee, H.S.; Lee, H.; Lee, D.H.; Lee, S.Y.; Hong, S. Melatonin ameliorates cerulein-induced pancreatitis by the modulation of nuclear erythroid 2-related factor 2 and nuclear factor-kappaB in rats. J. Pineal. Res. 2010, 48, 239–250. [Google Scholar] [CrossRef]
- Jung, C.H.; Kim, J.-H.; Park, S.; Kweon, D.-H.; Kim, S.-H.; Ko, S.-G. Inhibitory effect of Agrimonia pilosa Ledeb. on inflammation by suppression of iNOS and ROS production. Immunol. Investig. 2010, 39, 159–170. [Google Scholar] [CrossRef]
- Takahashi, T.; Kozaki, K.-I.; Yatabe, Y.; Achiwa, H.; Hida, T. Increased expression of COX-2 in the development of human lung cancers. J. Environ. Pathol. Toxicol. Oncol. 2002, 21, 5. [Google Scholar] [CrossRef]
- Vijayan, V. Heme oxygenase-1 as a therapeutic target in inflammatory disorders of the gastrointestinal tract. World J. Gastroenterol. 2010, 16, 3112–3119. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Antoniv, T.T.; Ji, J.D.; Ivashkiv, L.B. Lipopolysaccharide-induced expression of matrix metalloproteinases in human monocytes is suppressed by IFN-gamma via superinduction of ATF-3 and suppression of AP-1. J. Immunol. 2008, 181, 5089–5097. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.S.; Ramos, J.P.; Orozco, C.F.; Sánchez, M.A.Z.; Gutiérrez, S.P. Anti-Inflammatory Effects of Hyptis albida Chloroform Extract on Lipopolysaccharide-Stimulated Peritoneal Macrophages. ISRN Pharmacol. 2013, 2013, 713060. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, L.; Gao, L.; Ding, Q.; Yang, Q.; Kuai, J. Ulinastatin attenuates lipopolysaccharide‑induced cardiac dysfunction by inhibiting inflammation and regulating autophagy. Exp. Ther. Med. 2020, 20, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Moludi, J.; Maleki, V.; Jafari-Vayghyan, H.; Vaghef-Mehrabany, E.; Alizadeh, M. Metabolic endotoxemia and cardiovascular disease: A systematic review about potential roles of prebiotics and probiotics. Clin. Exp. Pharmacol. Physiol. 2020, 47, 927–939. [Google Scholar] [CrossRef]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.M.; Remouchamps, C.; McCorkell, K.A.; Solt, L.A.; Dejardin, E.; Orange, J.S.; May, M.J. Noncanonical NF-κB signaling is limited by classical NF-κB activity. Sci. Signal. 2014, 7, ra13. [Google Scholar] [CrossRef][Green Version]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Felices, M.J.; Escusol, S.; Martinez-Beamonte, R.; Gascón, S.; Barranquero, C.; Sanchez-De-Diego, C.; Osada, J.; Rodríguez-Yoldi, M.J. LPS-squalene interaction on d-galactose intestinal absorption. J. Physiol. Biochem. 2019, 75, 329–340. [Google Scholar] [CrossRef]
- Shih, T.-L.; Liu, M.-H.; Li, C.-W.; Kuo, C. Halo-Substituted Chalcones and Azachalcones Inhibited, Lipopolysaccharited-Stimulated, Pro-Inflammatory Responses through the TLR4-Mediated Pathway. Molecules 2018, 23, 597. [Google Scholar] [CrossRef] [PubMed]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac Injection of AdGRK5-NT Reduces Left Ventricular Hypertrophy by Inhibiting NF-κBDependent Hypertrophic Gene Expression. Hypertension 2010, 56, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Santulli, G.; Franco, A.; Cipolletta, E.; Napolitano, L.; Gambardella, J.; Gomez-Monterrey, I.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J. Cardiovasc. Transl. Res. 2015, 8, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-κB in the Heart. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Li, C.; Browder, W.; Kao, R.L. Early activation of transcription factor NF-kappaB during ischemia in perfused rat heart. Am. J. Physiol. 1999, 276, H543–H552. [Google Scholar]
- Li, C.; Kao, R.L.; Ha, T.; Kelley, J.; Browder, I.W.; Williams, D.L. Early activation of IKKbeta during in vivo myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, 1264–1271. [Google Scholar] [CrossRef]
- Valen, G.; Hansson, G.K.; Dumitrescu, A.; Vaage, J. Unstable angina activates myocardial heat shock protein 72, endothelial nitric oxide synthase, and transcription factors NFkappaB and AP-1. Cardiovasc. Res. 2000, 47, 49–56. [Google Scholar] [CrossRef]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory Cytokines and Postmyocardial Infarction Remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- Mann, D.L. Stress-Activated Cytokines and the Heart: From Adaptation to Maladaptation. Annu. Rev. Physiol. 2003, 65, 81–101. [Google Scholar] [CrossRef]
- Jang, J.C.; Li, J.; Gambini, L.; Batugedara, H.M.; Sati, S.; Lazar, M.A.; Fan, L.; Pellecchia, M.; Nair, M.G. Human resistin protects against endotoxic shock by blocking LPS–TLR4 interaction. Proc. Natl. Acad. Sci. USA 2017, 114, E10399–E10408. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Liu, X.-Y.; Han, G.; Wang, Z.-Y.; Li, X.-X.; Jiang, Z.-M.; Jiang, C.-M. LPS induces cardiomyocyte injury through calcium-sensing receptor. Mol. Cell. Biochem. 2013, 379, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.-Q. Expression of Toll-Like Receptors in Human Atherosclerotic Lesions. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Lv, X.; Sun, Y.; Ye, Z.; Kong, B.; Qin, Z. Role of TLR4/MyD88/NF-κB signaling pathway in coronary microembolization-induced myocardial injury prevented and treated with nicorandil. Biomed. Pharmacother. 2018, 106, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Kyoi, S.; Otani, H.; Matsuhisa, S.; Akita, Y.; Tatsumi, K.; Enoki, C.; Fujiwara, H.; Imamura, H.; Kamihata, H.; Iwasaka, T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 2006, 69, 888–898. [Google Scholar] [CrossRef]
- Sabapathy, K. Role of the JNK Pathway in Human Diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Flohé, L.; Brigelius-Flohé, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox Regulation of NF-kappa B Activation. Free. Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Vaziri, N. Causal link between oxidative stress, inflammation, and hypertension. Iran. J. Kidney Dis. 2008, 2, 1–10. [Google Scholar]
- Cachofeiro, V.; Goicochea, M.; De Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Blaha, M.J.; Blankstein, R.; Agatston, A.; Rivera, J.J.; Virani, S.S.; Ouyang, P.; Jones, S.R.; Blumenthal, R.S.; Budoff, M.J.; et al. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: Implications for statin therapy from the multi-ethnic study of atherosclerosis. Circulation 2014, 129, 77–86. [Google Scholar] [CrossRef]
- Leitinger, N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr. Opin. Lipidol. 2003, 14, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Mannarino, E.; Pirro, M. Molecular biology of atherosclerosis. Clin. Cases Miner. Bone Metab. 2008, 5, 57–62. [Google Scholar]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Orozco, L.D.; Kapturczak, M.H.; Barajas, B.; Wang, X.; Weinstein, M.M.; Wong, J.; Deshane, J.; Bolisetty, S.; Shaposhnik, Z.; Shih, D.M.; et al. Heme Oxygenase-1 Expression in Macrophages Plays a Beneficial Role in Atherosclerosis. Circ. Res. 2007, 100, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Heeba, G.H.; Moselhy, M.; Hassan, M.; Khalifa, M.; Gryglewski, R.; Malinski, T. Anti-atherogenic effect of statins: Role of nitric oxide, peroxynitrite and haem oxygenase-1. Br. J. Pharmacol. 2009, 156, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.A.; Vanhanen, H. Serum concentration and metabolism of cholesterol during rapeseed oil and squalene feeding. Am. J. Clin. Nutr. 1994, 59, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, T.E.; Tilvis, R.S.; Miettinen, T.A. Metabolic variables of cholesterol during squalene feeding in humans: Comparison with cholestyramine treatment. J. Lipid Res. 1990, 31, 1637–1643. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.N.M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Shin, J.-W.; Seol, I.; Son, C. Interpretation of Animal Dose and Human Equivalent Dose for Drug Development. J. Korean Orient. Med. 2010, 31, 1–7. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
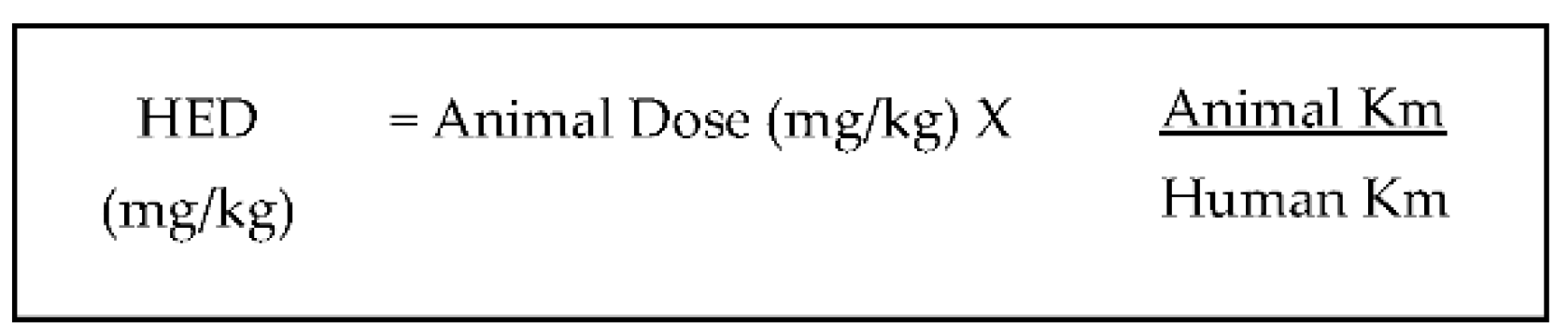{kind=link}
| Assays for Determining Antioxidant Activity | Cardiovascular-Related Conditions | Study Type | Experimental Model | Findings | Reference |
|---|---|---|---|---|---|
| Paraoxonase | Hyperlipidemia | Animal | Wild-type, ApoA1- and ApoE-deficient C57BL/6J mice | Reduction in reactive oxygen species (ROS) level and plasma malondialdehyde in lipoprotein fractions independently of the animal background. | [39] |
| Paraoxonase | Atherosclerosis | Animal | Female and male ApoE knockout mice | No significant changes in paraoxonase activity in both sexes. | [40] |
| 8-isoprostaglandin F2α | Decreased level of plasma 8-isoprostaglandin F2α in both sexes. | ||||
| Catalase (CAT) and superoxide dismutase (SOD) | Myocardial infarction (MI) | Animal | Isopreterenol MI-induced Wistar male rats | Increased CAT and SOD activities. Increased GPX and GST activities. | [41,45] |
| Glutathione peroxidase (GPX) and Glutathione-S-Transferase (GST) | |||||
| Glutathione (GSH) | Increased GSH. | [41,43,44] | |||
| Thiobarbituric Acid (TBARS) | Decreased lipid peroxidation in plasma and heart tissue. | [41,42,43,44] | |||
| GPx and GSH | Cardiotoxicity | Animal | Cyclophosphomide- induced cardiotoxicity in male Wistar rats | Increased GSH and decreased GPx. | [46] |
| Cardiovascular-Related Conditions | Study Type | Experimental Model | Findings | Reference |
|---|---|---|---|---|
| Atherosclerosis | In vitro | LPS-treated murine peritoneal macrophages | Suppression of iNOS and COX-2 protein expression. Significantly decreased phosphorylated JNK, but not p38 MAPK expression. Significantly decreased phosphorylated P65-NFκB, but significantly increase in IκB-α. Reduced mRNA levels of NFκB downstream genes including TNF- α and IL-1β. | [57] |
| LPS-treated human monocytes | Significantly downregulated TLR-4, iNOS and COX-2 gene expression. Significantly reduced pro-inflammatory cytokine genes, TNF-α and IL-1β, but not IL-6 or IL-10. Significantly downregulated MPO and upregulated anti-inflammatory gene HO-1. Significantly down-regulated MMP-1 and MMP-9 gene expression. Significantly upregulated PPARγ gene expression. | |||
| LPS-treated human neutrophils | Significantly downregulated TLR-4 and iNOS gene expression. Significantly reduced pro-inflammatory cytokine genes TNF-α, IL-1β, IL-6 and IFN-γ. Significantly downregulated MPO and upregulated anti-inflammatory gene HO-1. Significantly down-regulated MMP-1 and MMP-3 gene expression. Significantly upregulated PPARγ gene expression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, N.‘I.; Naina Mohamed, I. Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene–Implication for Cardiovascular Health. Life 2021, 11, 103. https://doi.org/10.3390/life11020103
Ibrahim N‘I, Naina Mohamed I. Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene–Implication for Cardiovascular Health. Life. 2021; 11(2):103. https://doi.org/10.3390/life11020103
Chicago/Turabian StyleIbrahim, Nurul ‘Izzah, and Isa Naina Mohamed. 2021. "Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene–Implication for Cardiovascular Health" Life 11, no. 2: 103. https://doi.org/10.3390/life11020103
APA StyleIbrahim, N. ‘I., & Naina Mohamed, I. (2021). Interdependence of Anti-Inflammatory and Antioxidant Properties of Squalene–Implication for Cardiovascular Health. Life, 11(2), 103. https://doi.org/10.3390/life11020103