
The Cell Protective Effect of Adenine on Hypoxia–Reoxygenation Injury through PPAR Delta Activation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Hypoxia–Reoxygenation of H9c2 Cells
2.3. Cell Number and Volume
2.4. Cell Viability Assay
2.5. Intracellular ROS/Superoxide Detection
2.6. Analysis of Glutathione (GSH)
2.7. Apoptosis and Cell Viability Assay
2.8. Quantitative RT-PCR
2.9. Western Blot Analysis
2.10. Antibodies
2.11. Statistical Analysis
3. Results
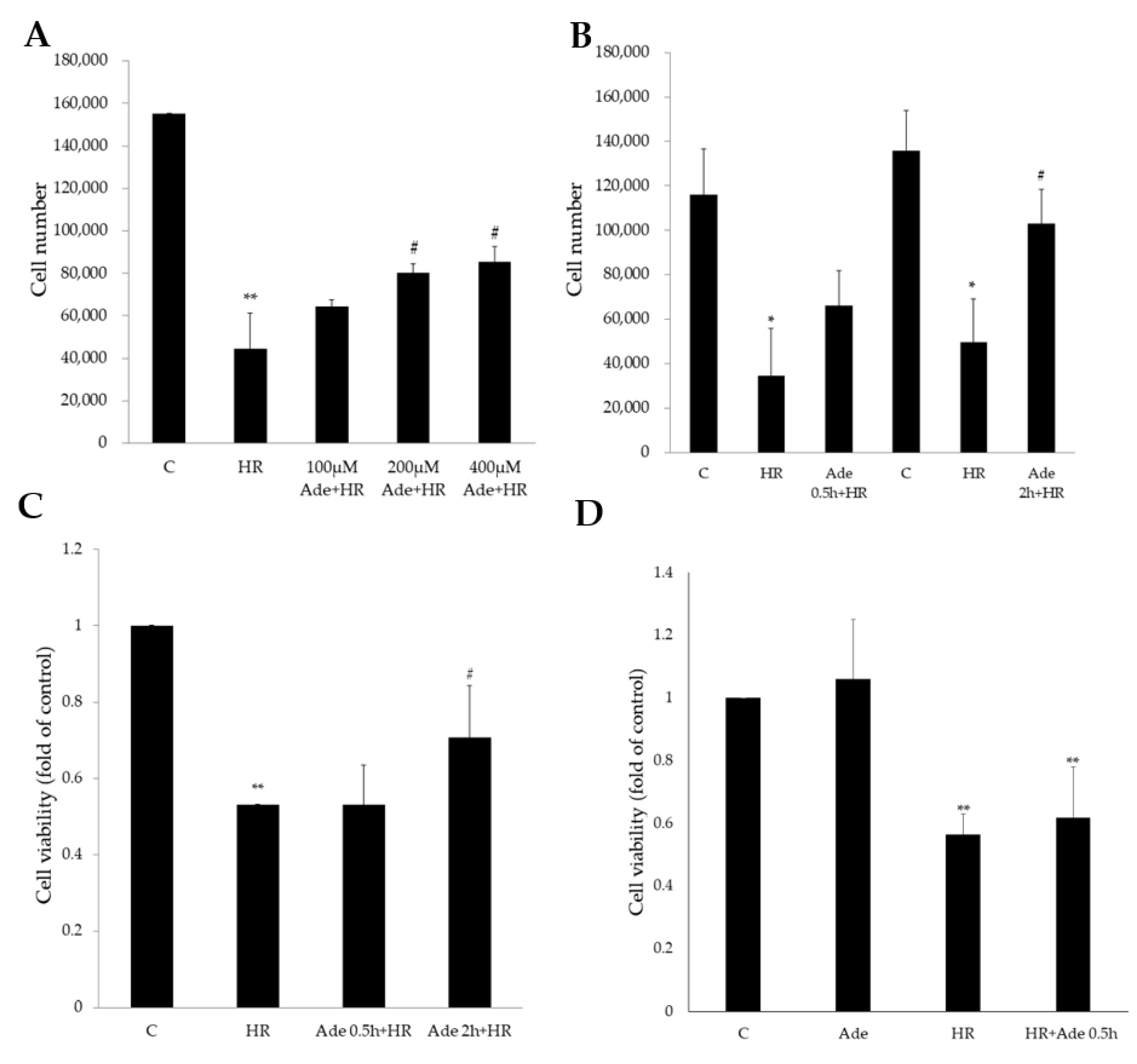
3.1. Protective Effect of Adenine in Hypoxia–Reoxygenation (HR) Cell Model
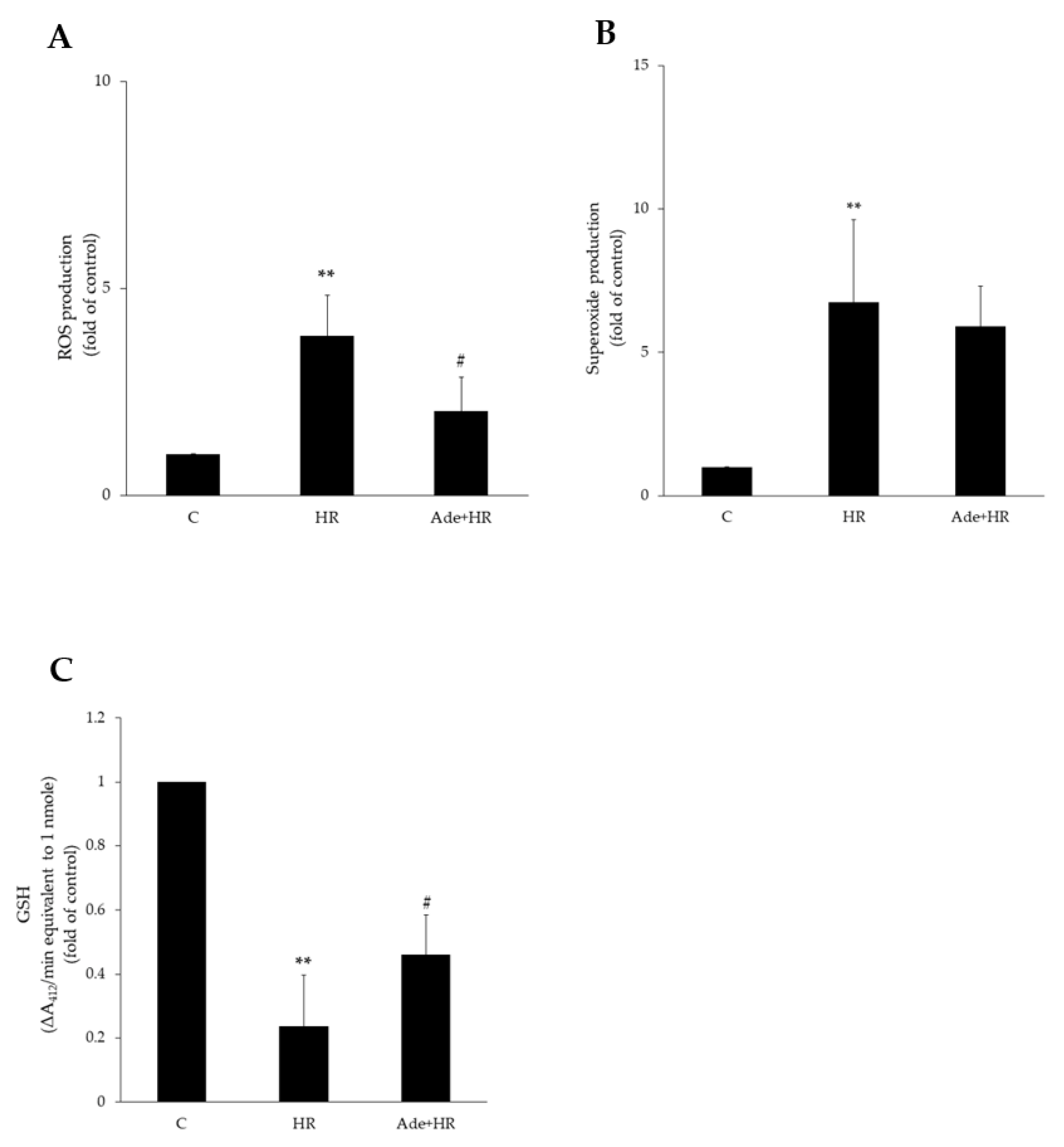
3.2. Antioxidant Effect of Adenine
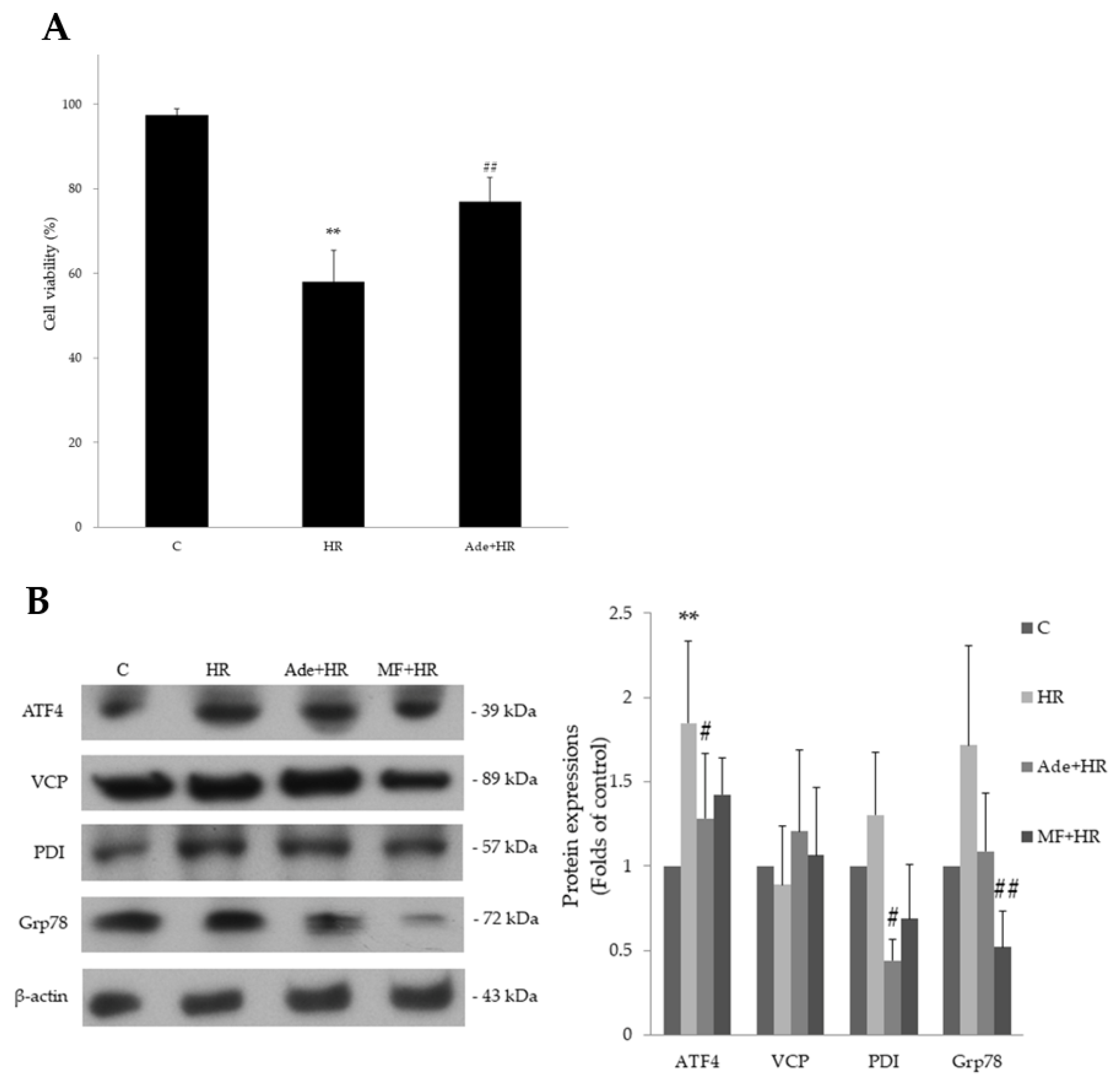
3.3. The Effect of Adenine on Cell Apoptosis after Hypoxia–Reoxygenation
3.4. The Effect of Adenine on Endoplasmic Reticulum Stress in HR Injury
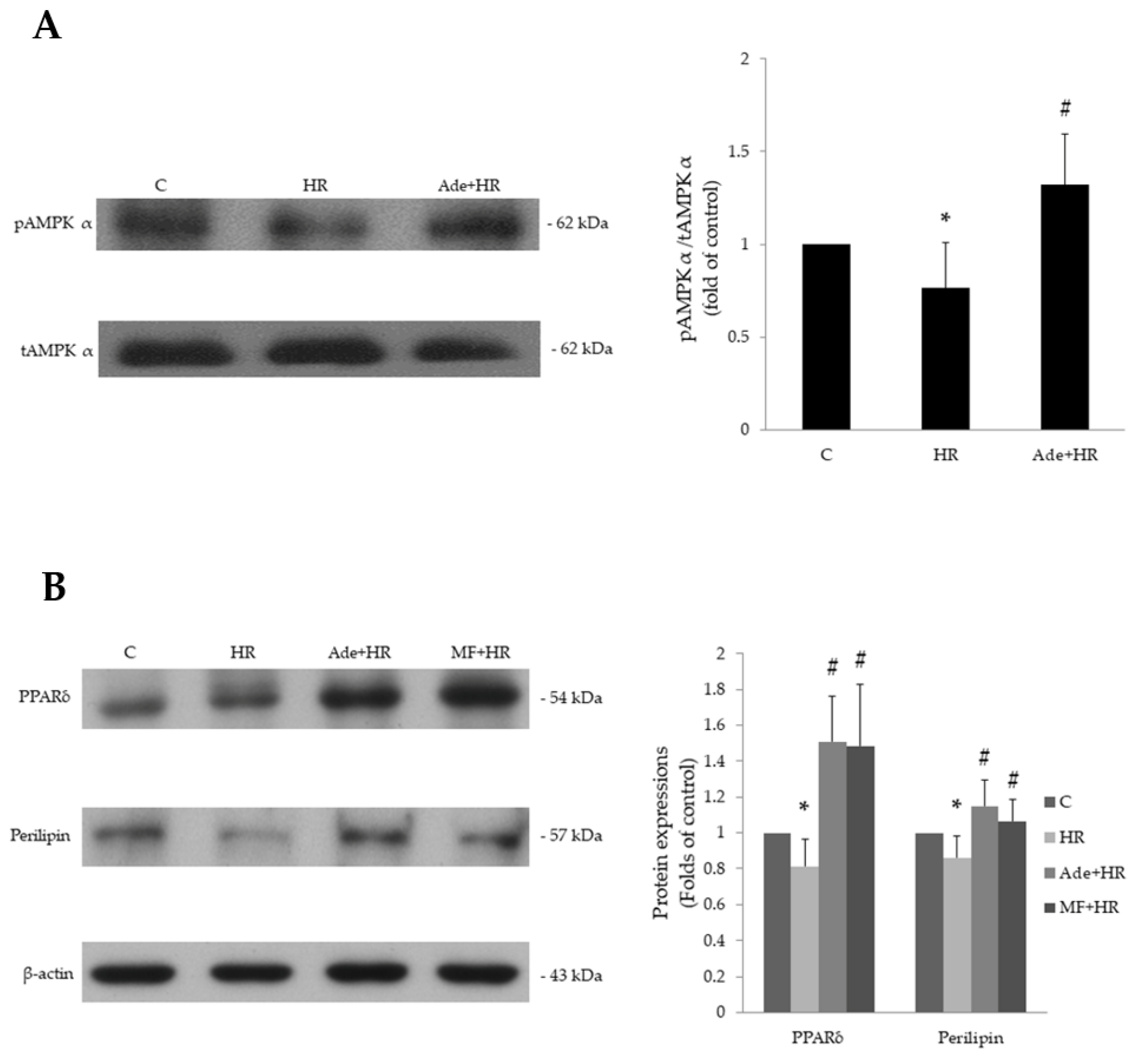
3.5. The Effect of Adenine on AMPK and PPARδ Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kandaswamy, E.; Zuo, L. Recent Advances in Treatment of Coronary Artery Disease: Role of Science and Technology. Int. J. Mol. Sci. 2018, 19, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doenst, T.; Haverich, A.; Serruys, P.; Bonow, R.O.; Kappetein, P.; Falk, V.; Velazquez, E.; Diegeler, A.; Sigusch, H. PCI and CABG for Treating Stable Coronary Artery Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF1alpha in myocardial ischemiareperfusion injury (Review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef] [PubMed]
- Garlick, P.B.; Davies, M.J.; Hearse, D.J.; Slater, T.F. Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ. Res. 1987, 61, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Nossuli, T.O.; Frangogiannis, N.G.; Knuefermann, P.; Lakshminarayanan, V.; Dewald, O.; Evans, A.J.; Peschon, J.; Mann, D.L.; Michael, L.H.; Entman, M.L. Brief murine myocardial I/R induces chemokines in a TNF-alpha-independent manner: Role of oxygen radicals. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2549–H2558. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef] [Green Version]
- De Villiers, C.; Riley, P.R. Mouse models of myocardial infarction: Comparing permanent ligation and ischaemia-reperfusion. Dis. Model Mech. 2020, 13, dmm046565. [Google Scholar] [CrossRef]
- Young, G.H.; Lin, J.T.; Cheng, Y.F.; Huang, C.F.; Chao, C.Y.; Nong, J.Y.; Chen, P.K.; Chen, H.M. Identification of adenine modulating AMPK activation in NIH/3T3 cells by proteomic approach. J. Proteom. 2015, 120, 204–214. [Google Scholar] [CrossRef]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Alexander, A.; Walker, C.L. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011, 585, 952–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Qiu, J.; Wang, X.; Zhang, Y.; Xia, M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2897–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zmijewski, J.W.; Lorne, E.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L497–L504. [Google Scholar] [CrossRef]
- Cheng, Y.F.; Young, G.H.; Lin, J.T.; Jang, H.H.; Chen, C.C.; Nong, J.Y.; Chen, P.K.; Kuo, C.Y.; Kao, S.H.; Liang, Y.J.; et al. Activation of AMP-Activated Protein Kinase by Adenine Alleviates TNF-Alpha-Induced Inflammation in Human Umbilical Vein Endothelial Cells. PLoS ONE 2015, 10, e0142283. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Youn, B.; Zheng, X.L.; Wu, D.; Xu, A.; Sweeney, G. Globular adiponectin, acting via AdipoR1/APPL1, protects H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS ONE 2011, 6, e19143. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.T.; Liang, Y.J.; Hsu, C.Y.; Chen, C.Y.; Chen, P.J.; Yang, Y.F.; Chen, Y.L.; Pei, D.; Chang, J.B.; Leu, J.G. Glucagon-like peptide receptor agonists attenuate advanced glycation end products-induced inflammation in rat mesangial cells. BMC Pharmacol. Toxicol. 2017, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Leu, J.G.; Su, W.H.; Chen, Y.C.; Liang, Y.J. Hydralazine attenuates renal inflammation in diabetic rats with ischemia/reperfusion acute kidney injury. Eur. J. Pharmacol. 2021, 910, 174468. [Google Scholar] [CrossRef]
- Zhu, H.; Zhou, H. Novel Insight into the Role of Endoplasmic Reticulum Stress in the Pathogenesis of Myocardial Ischemia-Reperfusion Injury. Oxid. Med. Cell Longev. 2021, 2021, 5529810. [Google Scholar] [CrossRef]
- Toral, M.; Jimenez, R.; Romero, M.; Robles-Vera, I.; Sanchez, M.; Salaices, M.; Sabio, J.M.; Duarte, J. Role of endoplasmic reticulum stress in the protective effects of PPARbeta/delta activation on endothelial dysfunction induced by plasma from patients with lupus. Arthritis Res. Ther. 2017, 19, 268. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Feng, J.; Tang, C.; Zhang, Z.; Feng, Y.; Duan, W.; Zhai, M.; Yan, Z.; Zhu, L.; Feng, L.; et al. Melatonin suppresses ER stress-dependent proapoptotic effects via AMPK in bone mesenchymal stem cells during mitochondrial oxidative damage. Stem Cell Res. Ther. 2020, 11, 442. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, G.; Zhou, W.; Ge, Y.; Fan, Z.; Liu, Q.; Gao, Y. Sestrin2 protects against bavachin induced ER stress through AMPK/mTORC1 signaling pathway in HepG2 cells. J. Pharmacol. Sci. 2021, 145, 175–186. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Peserico, D.; Stranieri, C.; Garbin, U.; Mozzini, C.C.; Danese, E.; Cominacini, L.; Pasini, F.A.M. Ezetimibe Prevents Ischemia/Reperfusion-Induced Oxidative Stress and Up-Regulates Nrf2/ARE and UPR Signaling Pathways. Antioxidants 2020, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Peng, H.; Wang, S.; Xu, X.; Xu, F.; Wang, Q.; Chen, Y.; Barton, L.A.; Chen, Y.; Zhang, Y.; et al. Mitochondrial ALDH2 protects against lipopolysaccharide-induced myocardial contractile dysfunction by suppression of ER stress and autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1627–1641. [Google Scholar] [CrossRef]
- Liao, J.; Sun, A.; Xie, Y.; Isse, T.; Kawamoto, T.; Zou, Y.; Ge, J. Aldehyde dehydrogenase-2 deficiency aggravates cardiac dysfunction elicited by endoplasmic reticulum stress induction. Mol. Med. 2012, 18, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-delta in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339. [Google Scholar] [CrossRef] [Green Version]
- Schubert, M.; Becher, S.; Wallert, M.; Maess, M.B.; Abhari, M.; Rennert, K.; Mosig, A.S.; Grosse, S.; Heller, R.; Grun, M.; et al. The Peroxisome Proliferator-Activated Receptor (PPAR)-gamma Antagonist 2-Chloro-5-Nitro-N-Phenylbenzamide (GW9662) Triggers Perilipin 2 Expression via PPARdelta and Induces Lipogenesis and Triglyceride Accumulation in Human THP-1 Macrophages. Mol. Pharmacol. 2020, 97, 212–225. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Zhang, L.; Zhang, M.; Li, L.; Luo, D.; Zhong, Y. Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic beta-cells. Nutr. Metab. 2019, 16, 50. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Tsai, T.H.; Li, L.; Saha, P.; Chan, L.; Chang, B.H. PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic beta Cells. Sci. Rep. 2017, 7, 40855. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Tang, W.; Zhang, Z.; Yang, C.; Qian, L.; Xie, X.; Qiang, E.; Zhao, J.; Zhao, W.; Xiao, L.; et al. Procyanidin B2 mitigates endothelial endoplasmic reticulum stress through a PPARdelta-Dependent mechanism. Redox. Biol. 2020, 37, 101728. [Google Scholar] [CrossRef] [PubMed]
- Tognola, C.; Alessandro, M.; Milani, M.; Cartella, I.; Tavecchia, G.; Grasso, E.; Sun, J.; Giannattasio, C. Nutraceuticals in Chronic Coronary Syndromes: Preclinical Data and Translational Experiences. High Blood Press. Cardiovasc. Prev. 2021, 28, 13–25. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leu, J.-G.; Wang, C.-M.; Chen, C.-Y.; Yang, Y.-F.; Shih, C.-Y.; Lin, J.-T.; Chen, H.-M.; Liang, Y.-J. The Cell Protective Effect of Adenine on Hypoxia–Reoxygenation Injury through PPAR Delta Activation. Life 2021, 11, 1408. https://doi.org/10.3390/life11121408
Leu J-G, Wang C-M, Chen C-Y, Yang Y-F, Shih C-Y, Lin J-T, Chen H-M, Liang Y-J. The Cell Protective Effect of Adenine on Hypoxia–Reoxygenation Injury through PPAR Delta Activation. Life. 2021; 11(12):1408. https://doi.org/10.3390/life11121408
Chicago/Turabian StyleLeu, Jyh-Gang, Chien-Mei Wang, Chao-Yi Chen, Yi-Feng Yang, Chin-Yu Shih, Jiun-Tsai Lin, Han-Min Chen, and Yao-Jen Liang. 2021. "The Cell Protective Effect of Adenine on Hypoxia–Reoxygenation Injury through PPAR Delta Activation" Life 11, no. 12: 1408. https://doi.org/10.3390/life11121408
APA StyleLeu, J.-G., Wang, C.-M., Chen, C.-Y., Yang, Y.-F., Shih, C.-Y., Lin, J.-T., Chen, H.-M., & Liang, Y.-J. (2021). The Cell Protective Effect of Adenine on Hypoxia–Reoxygenation Injury through PPAR Delta Activation. Life, 11(12), 1408. https://doi.org/10.3390/life11121408