
LoopTag FRET Probe System for Multiplex qPCR Detection of Borrelia Species

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. qPCR Reaction and Melting Curve Analysis
2.2. Borrelia Strains and DNA Extraction and qPCR Conditions
Data Analysis
3. Results
3.1. Mechanism of the LoopTag System
3.2. Amplification of the Flagellin Gene
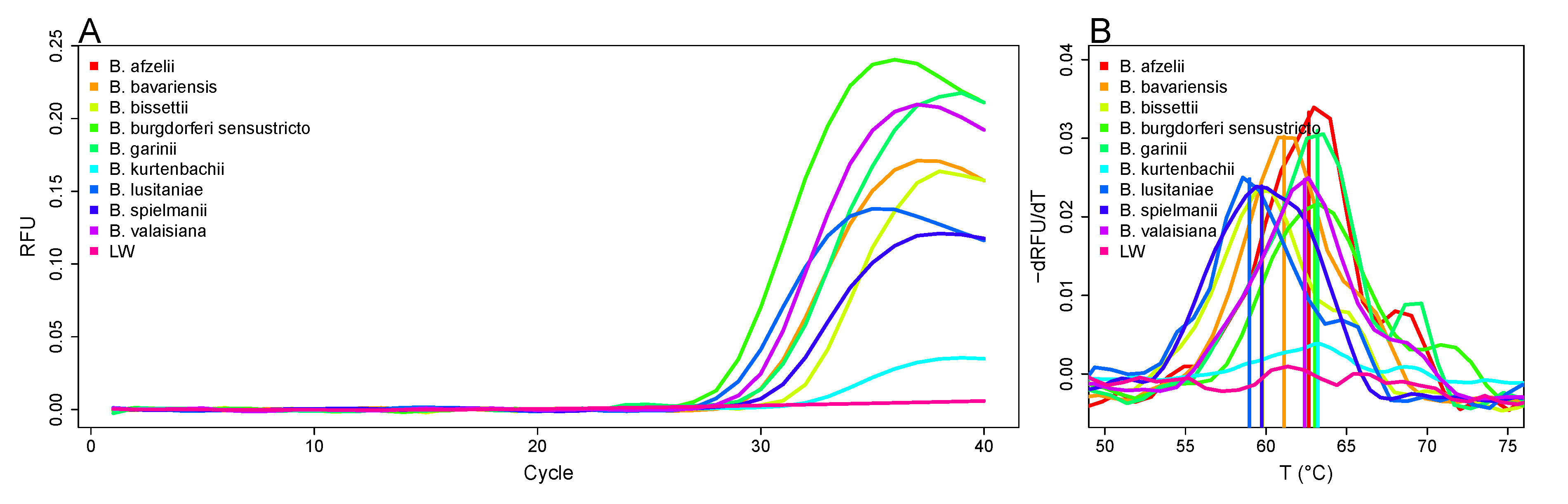
3.3. Differentiation of Borrelia Species Based on Melting Curve Analysis
3.4. Internal Control for the Detection and Differentiation of Borrelia Species
3.5. Sensitivity and Efficiency
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A.U. | Arbitrary units |
| BHQ2 | Black Hole Quencher 2 |
| Cq | Cycle of quantification by SDM method |
| estA1 | Heat-stable toxin I gene |
| FRET | Foerster Resonance Energy Transfer |
| qPCR | Quantitative PCR |
| RFU | Relative fluorescence units [Arbitrary Unit] |
| rMFI | Reference mean fluorescence value (see [47]) |
| SDM | Second derivative maximum (see [42,48]) |
| stx1 | Shigatoxin 1 gene |
| Tm | Melting temperature |
Appendix A. Melting Point Analysis of Borrelia Specific Probes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Difference (°C) | Lower | Upper | p |
|---|---|---|---|---|
| B. bavariensis vs. B. afzelii | −1.55 | −1.98 | −1.12 | <0.001 |
| B. bissettii vs. B. afzelii | −2.70 | −3.13 | −2.27 | <0.001 |
| B. burgdorferi vs. B. afzelii | 0.46 | 0.02 | 0.89 | 0.03 |
| B. lusitaniae vs. B. afzelii | −3.48 | −3.91 | −3.04 | <0.001 |
| B. spielmanii vs. B. afzelii | −4.24 | −4.67 | −3.81 | <0.001 |
| B. bissettii vs. B. bavariensis | −1.15 | −1.58 | −0.72 | <0.001 |
| B. burgdorferi vs. B. bavariensis | 2.00 | 1.57 | 2.44 | <0.001 |
| B. garinii vs. B. bavariensis | 1.56 | 1.12 | 1.99 | <0.001 |
| B. kurtenbachii vs. B. bavariensis | 1.52 | 1.09 | 1.96 | <0.001 |
| B. lusitaniae vs. B. bavariensis | −1.93 | −2.36 | −1.50 | <0.001 |
| B. spielmanii vs. B. bavariensis | −2.69 | −3.12 | −2.26 | <0.001 |
| B. valaisiana vs. B. bavariensis | 1.79 | 1.35 | 2.22 | <0.001 |
| B. burgdorferi vs. B. bissettii | 3.15 | 2.72 | 3.59 | <0.001 |
| B. garinii vs. B. bissettii | 2.71 | 2.27 | 3.14 | <0.001 |
| B. kurtenbachii vs. B. bissettii | 2.68 | 2.24 | 3.11 | <0.001 |
| B. lusitaniae vs. B. bissettii | −0.78 | −1.21 | −0.35 | <0.001 |
| B. spielmanii vs. B. bissettii | −1.54 | −1.97 | −1.11 | <0.001 |
| B. valaisiana vs. B. bissettii | 2.94 | 2.50 | 3.37 | <0.001 |
| B. garinii vs. B. burgdorferi | −0.45 | −0.88 | −0.01 | 0.04 |
| B. kurtenbachii vs. B. burgdorferi | −0.48 | −0.91 | −0.05 | 0.02 |
| B. lusitaniae vs. B. burgdorferi | −3.93 | −4.37 | −3.50 | <0.001 |
| B. spielmanii vs. B. burgdorferi | −4.69 | −5.13 | −4.26 | <0.001 |
| B. lusitaniae vs. B. garinii | −3.49 | −3.92 | −3.05 | <0.001 |
| B. spielmanii vs. B. garinii | −4.25 | −4.68 | −3.81 | <0.001 |
| B. lusitaniae vs. B. kurtenbachii | −3.45 | −3.89 | −3.02 | <0.001 |
| B. spielmanii vs. B. kurtenbachii | −4.22 | −4.65 | −3.78 | <0.001 |
| B. spielmanii vs. B. lusitaniae | −0.76 | −1.20 | −0.33 | <0.001 |
| B. valaisiana vs. B. lusitaniae | 3.71 | 3.28 | 4.15 | <0.001 |
| B. valaisiana vs. B. spielmanii | 4.48 | 4.04 | 4.91 | <0.001 |
Appendix B. Duplex LoopTag qPCR on a Planar Microbead Assays

- Base primer sequences were taken from [49] and transformed into a LoopTag probe system as described above. An important difference is that a part of the probes are bound to the surface of microbeads, so that they can function as catcher molecules (Table A2). The gene estA1 (length of amplification product: 158 bp) and stx1 (length of amplification product: 211 bp) were used in this proof-of-concept study.
- Colonies of E. coli strains containing genes estA1 and stx1 were inoculated in 1.5 mL LB medium. After incubation at 37 °C overnight cells were separated, re-suspended in 300 L of water, and lysed by heating at 99 °C for 10 min. Cell debris were removed by centrifugation and supernatant was stored at −20 °C. Thawed lysates were directly used as templates for PCR.
| Function | Sequence |
|---|---|
| estA1 fw | BHQ2-ATCTACCAACTGAATCACTTGACTCTT (GC: 37%, 27 nt, Tm: 62.45 °C) |
| estA1 rv | TTAATAACATCCAGCACAGG (GC: 40%, 20 nt, Tm: 55.57 °C) |
| estA1 probe | 2B-AGTCTCTAATGTAATTTTCTCTTTTGGTAGAT-Atto647N (GC: 28%, 32 nt, Tm: 61.51 °C) |
| stx1 fw | BHQ2-ATGTATGTTGCAGGGATCAGTCGT (GC: 45%, 24 nt, Tm: 64.57 °C) |
| stx1 rv | AGAACGCCCACTGAGATCATC (GC: 52%, 21 nt, Tm: 62.41 °C) |
| stx1 probe | 2B-GTCAACGAATGGCGATTTATCTGCATCCCGTACAT-Atto647N (GC: 45%, 35 nt, Tm: 62.41 °C) |
- Briefly, 300,000 microbeads were re-suspended in 100 L of 100 mM methylimidazole (MeIm, Sigma, Milwaukee, WI, USA) buffer (pH 7.0) containing 25 mg/mL N-(3-dimethy-laminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, Sigma). Activated microbeads were incubated with streptavidin solution (300 g/mL in MeIm-buffer for 5 h at 50 °C) with continuous agitation for covalent cross-linking. After washing three times with TBST buffer, streptavidin-coated microbeads were ready for loading with biotinylated oligonucleotides. Microbeads were mixed with 100 L TBST buffer containing 100 nM of 5-bis-biotinylated and 3-Atto647N-labeled oligonucleotides (biomers.net, Ulm, Germany). After 15 min incubation at room temperature, unbound oligonucleotides were removed by washing microbeads three times with 200 L TBST. Finally, microbeads were exposed to 95 °C for 10 min.
- All PCR reactions were conducted within a volume of 20 L Biotherm polymerase buffer (Genecraft, Colone, Germany) containing 5 mM MgCl, 200 M of each dNTP, 250 M of BHQ2 labeled fw primer, 250 M of rv primer, 20–200 streptavidin-coupled microbeads with bound bis-biotinylated capture probes, 1 U Biotherm DNA-polymerase (Genecraft), and 1 L of E. coli lysate as template. The mixture was transferred into a cavity of a nucleolink TopYield strip (Nunc, Roskilde, Denmark), covered with 30 L of mineral oil, and the strip was placed into sockets of the heating and cooling unit. Here, a PCR was performed using the following program: 4 min, 94 °C, 40× (60 s 94 °C, 60 s 55 °C, 90 s 72 °C), 5 min 72 °C. We monitored microbead surface fluorescence while the temperature was increased at 1 °C/min starting from 35 °C to 90 °C. By use of the VideoScan technology fluorescence images of microbeads on the bottom of the well were taken after every cycle (55 °C). Images were evaluated with imaging processing software allowing the recognition of microbeads, the assignment to a population and the determination of their surface fluorescence.
- The mean fluorescence value for every population (rMFI → RFU) was plotted against the cycle number leading to the shown real-time kinetic curve. The amplification curve data were pre-processed and analyzed using the qpcR (v. 1.4.1) [50], MBmca, and chipPCR packages. After finishing real-time duplex PCR, a melting curve analysis was done as described in [39,43].
References
- Hildebrandt, A.; Schmidt, K.H.; Wilske, B.; Dorn, W.; Straube, E.; Fingerle, V. Prevalence of four species of Borrelia burgdorferi sensu lato and coinfection with Anaplasma phagocytophila in Ixodes ricinus ticks in central Germany. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2003, 22, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Park, H.S.; Jang, W.J.; Koh, S.E.; Yang, Y.M.; Kim, B.J.; Kook, Y.H.; Park, K.H. Differentiation of Borrelia burgdorferi sensu lato through groEL gene analysis. FEMS Microbiol. Lett. 2003, 222, 51–57. [Google Scholar] [CrossRef]
- Knoll, S.; Springer, A.; Hauck, D.; Schunack, B.; Pachnicke, S.; Fingerle, V.; Strube, C. Distribution of Borrelia burgdorferi s.l. and Borrelia miyamotoi in Ixodes tick populations in Northern Germany, co-infections with Rickettsiales and assessment of potential influencing factors. Med. Vet. Entomol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tappe, J.; Jordan, D.; Janecek, E.; Fingerle, V.; Strube, C. Revisited: Borrelia burgdorferi sensu lato infections in hard ticks (Ixodes ricinus) in the city of Hanover (Germany). Parasites Vectors 2014, 7, 441. [Google Scholar] [CrossRef]
- Răileanu, C.; Tauchmann, O.; Vasić, A.; Wöhnke, E.; Silaghi, C. Borrelia miyamotoi and Borrelia burgdorferi (sensu lato) identification and survey of tick-borne encephalitis virus in ticks from north-eastern Germany. Parasites Vectors 2020, 13, 106. [Google Scholar] [CrossRef]
- Mendoza-Roldan, J.A.; Colella, V.; Lia, R.P.; Nguyen, V.L.; Barros-Battesti, D.M.; Iatta, R.; Dantas-Torres, F.; Otranto, D. Borrelia burgdorferi (sensu lato) in ectoparasites and reptiles in southern Italy. Parasites Vectors 2019, 12, 35. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Schmidt, K.H.; Fingerle, V.; Wilske, B.; Straube, E. Prevalence of granulocytic Ehrlichiae in Ixodes ricinus ticks in Middle Germany (Thuringia) detected by PCR and sequencing of a 16S ribosomal DNA fragment. FEMS Microbiol. Lett. 2002, 211, 225–230. [Google Scholar] [CrossRef]
- Deutschmann, C.; Roggenbuck, D.; Schierack, P.; Rödiger, S. Autoantibody testing by enzyme-linked immunosorbent assay—A case in which the solid phase decides on success and failure. Heliyon 2020, 6, e03270. [Google Scholar] [CrossRef]
- Lager, M.; Faller, M.; Wilhelmsson, P.; Kjelland, V.; Andreassen, A.; Dargis, R.; Quarsten, H.; Dessau, R.; Fingerle, V.; Margos, G.; et al. Molecular detection of Borrelia burgdorferi sensu lato—An analytical comparison of real-time PCR protocols from five different Scandinavian laboratories. PLoS ONE 2017, 12, e0185434. [Google Scholar] [CrossRef]
- Primus, S.; Akoolo, L.; Schlachter, S.; Gedroic, K.; Rojtman, A.D.; Parveen, N. Efficient detection of symptomatic and asymptomatic patient samples for Babesia microti and Borrelia burgdorferi infection by multiplex qPCR. PLoS ONE 2018, 13, e0196748. [Google Scholar] [CrossRef]
- Venczel, R.; Knoke, L.; Pavlovic, M.; Dzaferovic, E.; Vaculova, T.; Silaghi, C.; Overzier, E.; Konrad, R.; Kolenčík, S.; Derdakova, M.; et al. A novel duplex real-time PCR permits simultaneous detection and differentiation of Borrelia miyamotoi and Borrelia burgdorferi sensu lato. Infection 2016, 44, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Alby, K.; Capraro, G.A. Alternatives to Serologic Testing for Diagnosis of Lyme Disease. Clin. Lab. Med. 2015, 35, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Pabinger, S.; Rödiger, S.; Kriegner, A.; Vierlinger, K.; Weinhäusel, A. A survey of tools for the analysis of quantitative PCR (qPCR) data. Biomol. Detect. Quantif. 2014, 1, 23–33. [Google Scholar] [CrossRef]
- Maurin, M. Real-time PCR as a diagnostic tool for bacterial diseases. Expert Rev. Mol. Diagn. 2012, 12, 731–754. [Google Scholar] [CrossRef]
- Saiki, R.K.; Bugawan, T.L.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature 1986, 324, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.L. PCR in laboratory diagnosis of human Borrelia burgdorferi infections. Clin. Microbiol. Rev. 1997, 10, 185–201. [Google Scholar] [CrossRef]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain reaction product by utilizing the 5′–3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR Analysis: Real-time Monitoring of DNA Amplification Reactions. Nat. Biotechnol. 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Lorenz, P.; Tuomi, J.M.; Hecker, M.; Hoff, M.J.B.V.D. Fluorescent-increase kinetics of different fluorescent reporters used for qPCR depend on monitoring chemistry, targeted sequence, type of DNA input and PCR efficiency. Microchim. Acta 2014, 181, 1689–1696. [Google Scholar] [CrossRef]
- Forootan, A.; Sjöback, R.; Björkman, J.; Sjögreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Navarro, E.; Serrano-Heras, G.; Castano, M.J.; Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta 2015, 439, 231–250. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Thelwell, N.; Millington, S.; Solinas, A.; Booth, J.; Brown, T. Mode of action and application of Scorpion primers to mutation detection. Nucleic Acids Res. 2000, 28, 3752–3761. [Google Scholar] [CrossRef]
- Rödiger, S.; Liebsch, C.; Schmidt, C.; Lehmann, W.; Resch-Genger, U.; Schedler, U.; Schierack, P. Nucleic acid detection based on the use of microbeads: A review. Microchim. Acta 2014, 181, 1151–1168. [Google Scholar] [CrossRef]
- Ranasinghe, R.T.; Brown, T. Fluorescence based strategies for genetic analysis. Chem. Commun. (Camb. Engl.) 2005, 44, 5487–5502. [Google Scholar] [CrossRef]
- Bernard, P.S.; Ajioka, R.S.; Kushner, J.P.; Wittwer, C.T. Homogeneous multiplex genotyping of hemochromatosis mutations with fluorescent hybridization probes. Am. J. Pathol. 1998, 153, 1055–1061. [Google Scholar] [CrossRef]
- Lay, M.J.; Wittwer, C.T. Real-time fluorescence genotyping of factor V Leiden during rapid-cycle PCR. Clin. Chem. 1997, 43, 2262–2267. [Google Scholar] [CrossRef] [PubMed]
- Marras, S.A.; Kramer, F.R.; Tyagi, S. Multiplex detection of single-nucleotide variations using molecular beacons. Genet. Anal. Biomol. Eng. 1999, 14, 151–156. [Google Scholar] [CrossRef]
- Schwaiger, M.; Péter, O.; Cassinotti, P. Routine diagnosis of Borrelia burgdorferi (sensu lato) infections using a real-time PCR assay. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2001, 7, 461–469. [Google Scholar] [CrossRef]
- Lehmann, W.; Hanschmann, H.; Syring, M. Method and Probe/Primer System for the “Real Time” Detection of a Nucleic Acid Target. International Patent WO2008152144A1, 13 June 2008. [Google Scholar]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Marshall, O. Graphical Design of Primers with PerlPrimer. In PCR Primer Design; Yuryev, A., Ed.; Number 402 in Methods in Molecular Biology™; Humana Press: Totowa, NJ, USA, 2007; pp. 403–414. [Google Scholar] [CrossRef]
- Ponti, G.; Maccaferri, M.; Manfredini, M.; Kaleci, S.; Mandrioli, M.; Pellacani, G.; Ozben, T.; Depenni, R.; Bianchi, G.; Pirola, G.M.; et al. The value of fluorimetry (Qubit) and spectrophotometry (NanoDrop) in the quantification of cell-free DNA (cfDNA) in malignant melanoma and prostate cancer patients. Clin. Chim. Acta 2018, 479, 14–19. [Google Scholar] [CrossRef]
- Wright, K.; de Silva, K.; Purdie, A.C.; Plain, K.M. Comparison of methods for miRNA isolation and quantification from ovine plasma. Sci. Rep. 2020, 10, 825. [Google Scholar] [CrossRef]
- Nakayama, Y.; Yamaguchi, H.; Einaga, N.; Esumi, M. Pitfalls of DNA Quantification Using DNA-Binding Fluorescent Dyes and Suggested Solutions. PLoS ONE 2016, 11, e0150528. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Leung, W.Y.; Xin, X. Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 2007, 7, 76. [Google Scholar] [CrossRef]
- Rödiger, S.; Friedrichsmeier, T.; Kapat, P.; Michalke, M. RKWard: A comprehensive graphical user interface and integrated development environment for statistical analysis with R. J. Stat. Softw. 2012, 49, 1–34. [Google Scholar] [CrossRef]
- Rödiger, S.; Burdukiewicz, M.; Blagodatskikh, K.A.; Schierack, P. R as an Environment for the Reproducible Analysis of DNA Amplification Experiments. R J. 2015, 7, 127–150. [Google Scholar] [CrossRef]
- Makowski, D.; Ben-Shachar, M.S.; Patil, I.; Lüdecke, D. Automated Results Reporting as a Practical Tool to Improve Reproducibility and Methodological Best Practices Adoption; Technical Report; Easystats: Rocklin, CA, USA, 2021. [Google Scholar]
- Rödiger, S.; Burdukiewicz, M.; Schierack, P. chipPCR: An R package to pre-process raw data of amplification curves. Bioinformatics 2015, 31, 2900–2902. [Google Scholar] [CrossRef] [PubMed]
- Spiess, A.N.; Deutschmann, C.; Burdukiewicz, M.; Himmelreich, R.; Klat, K.; Schierack, P.; Rödiger, S. Impact of Smoothing on Parameter Estimation in Quantitative DNA Amplification Experiments. Clin. Chem. 2015, 61, 379–388. [Google Scholar] [CrossRef]
- Rödiger, S.; Böhm, A.; Schimke, I. Surface Melting Curve Analysis with R. R J. 2013, 5, 37–53. [Google Scholar] [CrossRef]
- Aguero-Rosenfeld, M.E.; Wang, G.; Schwartz, I.; Wormser, G.P. Diagnosis of Lyme Borreliosis. Clin. Microbiol. Rev. 2005, 18, 484–509. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Müller, T.; Fingerle, V.; Noll, M. Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations. Microorganisms 2021, 9, 1814. [Google Scholar] [CrossRef]
- Burdukiewicz, M.; Spiess, A.N.; Blagodatskikh, K.A.; Lehmann, W.; Schierack, P.; Rödiger, S. Algorithms for automated detection of hook effect-bearing amplification curves. Biomol. Detect. Quantif. 2018, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Rödiger, S.; Schierack, P.; Böhm, A.; Nitschke, J.; Berger, I.; Frömmel, U.; Schmidt, C.; Ruhland, M.; Schimke, I.; Roggenbuck, D.; et al. A highly versatile microscope imaging technology platform for the multiplex real-time detection of biomolecules and autoimmune antibodies. Adv. Biochem. Eng. 2013, 133, 35–74. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Ruijter, J.M.; van den Hoff, M.J.B. Use and Misuse of Cq in qPCR Data Analysis and Reporting. Life 2021, 11, 496. [Google Scholar] [CrossRef]
- Frömmel, U.; Lehmann, W.; Rödiger, S.; Böhm, A.; Nitschke, J.; Weinreich, J.; Groß, J.; Roggenbuck, D.; Zinke, O.; Ansorge, H.; et al. Adhesion of Human and Animal Escherichia coli Strains in Association with Their Virulence-Associated Genes and Phylogenetic Origins. Appl. Environ. Microbiol. 2013, 79, 5814–5829. [Google Scholar] [CrossRef]
- Ritz, C.; Spiess, A.N. qpcR: An R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics 2008, 24, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Spiess, A.N.; Rödiger, S.; Burdukiewicz, M.; Volksdorf, T.; Tellinghuisen, J. System-specific periodicity in quantitative real-time polymerase chain reaction data questions threshold-based quantitation. Sci. Rep. 2016, 6, 38951. [Google Scholar] [CrossRef]
- Schneider, J.; Weiss, R.; Ruhe, M.; Jung, T.; Roggenbuck, D.; Stohwasser, R.; Schierack, P.; Rödiger, S. Open source bioimage informatics tools for the analysis of DNA damage and associated biomarkers. J. Lab. Precis. Med. 2019, 4, 1–27. [Google Scholar] [CrossRef]




| Species | Melting Temperature (°C) |
|---|---|
| B. afzelii | 62.4 ± 0.1 |
| B. bavariensis | 61.1 ± 0.4 |
| B. bissettii | 60.2 ± 0.8 |
| B. burgdorferii | 63.7 ± 0.7 |
| B. garinii | 62.3 ± 0.3 |
| B. kurtenbachii | 63.6 ± 0.3 |
| B. lusitaniae | 59.7 ± 1 |
| B. spielmanii | 59.1 ± 0.5 |
| B. valaisiana | 62.4 ± 0.2 |
| Borrelia | Species | DMAGE |
|---|---|---|
| Borrelia burgdorferi s.l. complex | B. afzelii | ≥10 |
| B. bavariensis | ≥10 | |
| B. bissettii | ≥10 | |
| B. burgdorferi s.s. | ≥10 | |
| B. garinii | PBr : approx. 10 | |
| PHei: ≥10 | ||
| TN: approx. 700 | ||
| PRef: ≥10 | ||
| PLa: ≥10 | ||
| PWudII: ≥10 | ||
| B. kurtenbachii | 100 | |
| B. lusitaniae | ≥10 | |
| B. spielmanii | ≥10 | |
| B. valaisiana | ≥10 | |
| Relapsing fever Borrelia | B. anserina | ≥400.000 * |
| B. duttonii | ≥400.000 * | |
| B. miyamotoi | Not detectable | |
| B. parkerii | ≥400.000 * | |
| B. recurrentis | Not detectable | |
| B. turicatae | ≥200.000 * | |
| Negative controls (other species) | E. coli (2 strains) | Not detectable |
| Leptospira (2 strains) | Not detectable | |
| Treponema phagedenis (2 strains) | Not detectable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanschmann, H.; Rödiger, S.; Kramer, T.; Hanschmann, K.; Steidle, M.; Fingerle, V.; Schmidt, C.; Lehmann, W.; Schierack, P. LoopTag FRET Probe System for Multiplex qPCR Detection of Borrelia Species. Life 2021, 11, 1163. https://doi.org/10.3390/life11111163
Hanschmann H, Rödiger S, Kramer T, Hanschmann K, Steidle M, Fingerle V, Schmidt C, Lehmann W, Schierack P. LoopTag FRET Probe System for Multiplex qPCR Detection of Borrelia Species. Life. 2021; 11(11):1163. https://doi.org/10.3390/life11111163
Chicago/Turabian StyleHanschmann, Henning, Stefan Rödiger, Toni Kramer, Katrin Hanschmann, Michael Steidle, Volker Fingerle, Carsten Schmidt, Werner Lehmann, and Peter Schierack. 2021. "LoopTag FRET Probe System for Multiplex qPCR Detection of Borrelia Species" Life 11, no. 11: 1163. https://doi.org/10.3390/life11111163
APA StyleHanschmann, H., Rödiger, S., Kramer, T., Hanschmann, K., Steidle, M., Fingerle, V., Schmidt, C., Lehmann, W., & Schierack, P. (2021). LoopTag FRET Probe System for Multiplex qPCR Detection of Borrelia Species. Life, 11(11), 1163. https://doi.org/10.3390/life11111163