
Molecular Analysis of Vietnamese Patients with Mucopolysaccharidosis Type I

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients, Clinical Data and Biochemical Profiles
2.2. Molecular Genetic Analysis
3. Results
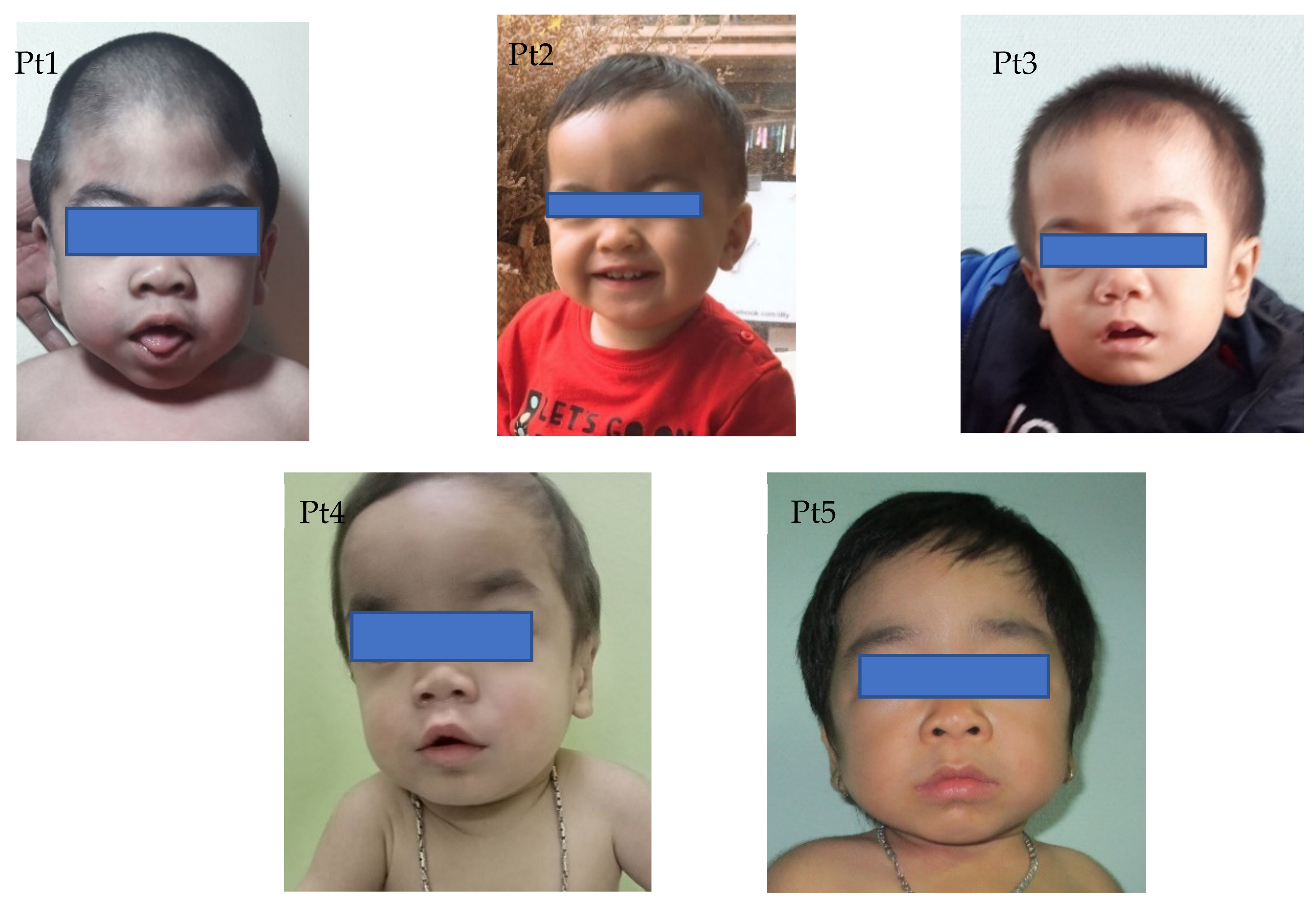
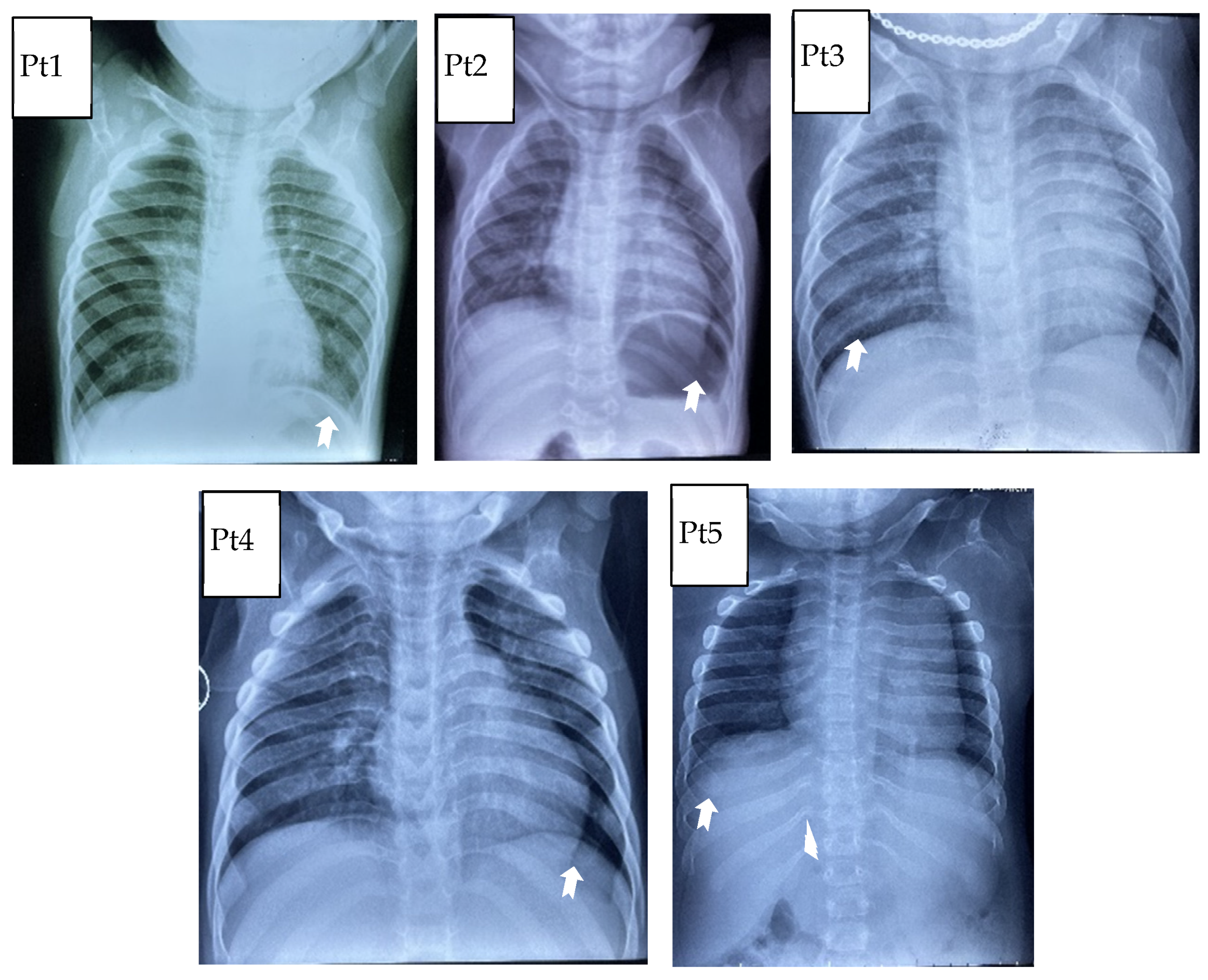
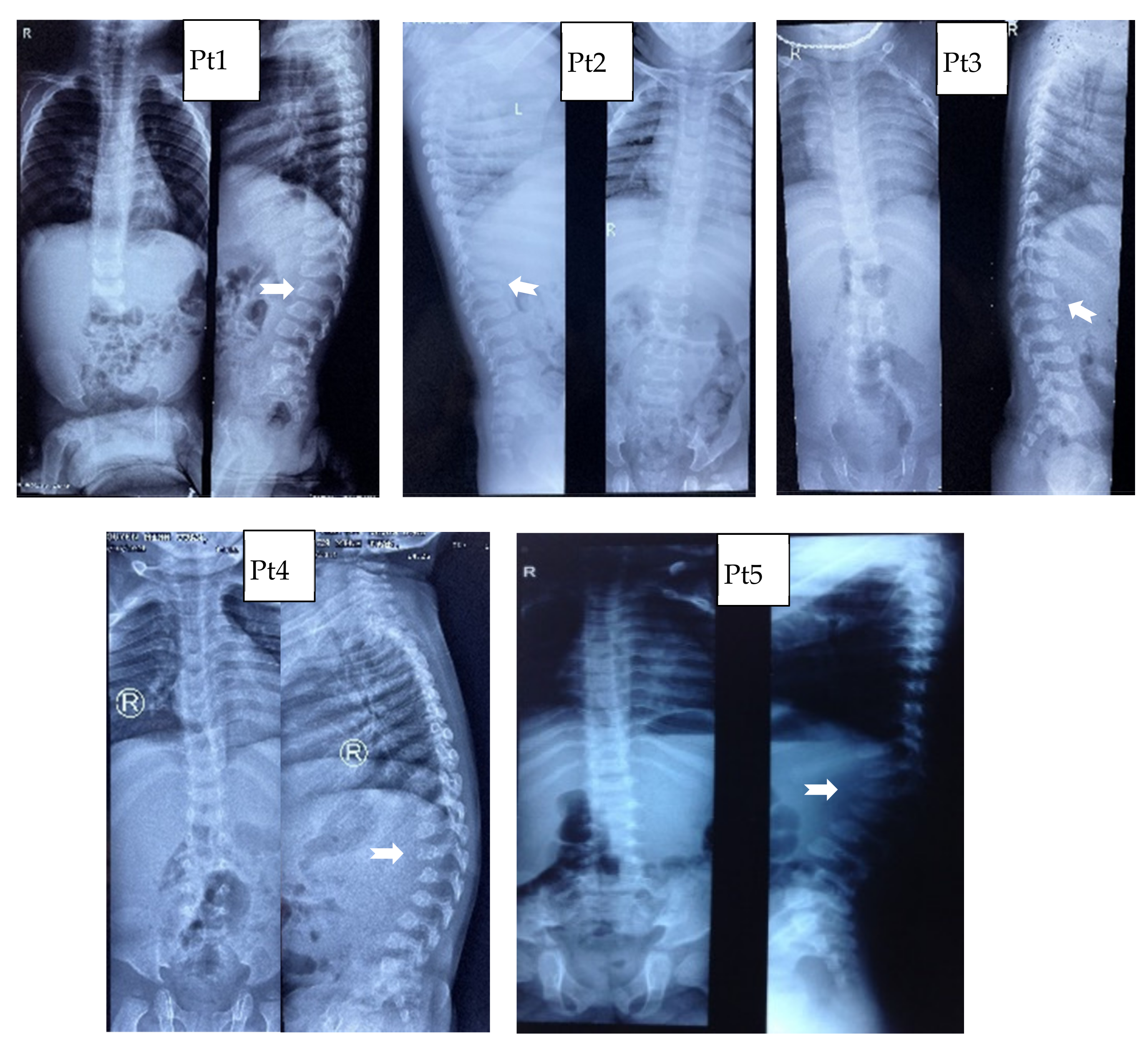
3.1. Clinical Characteristics
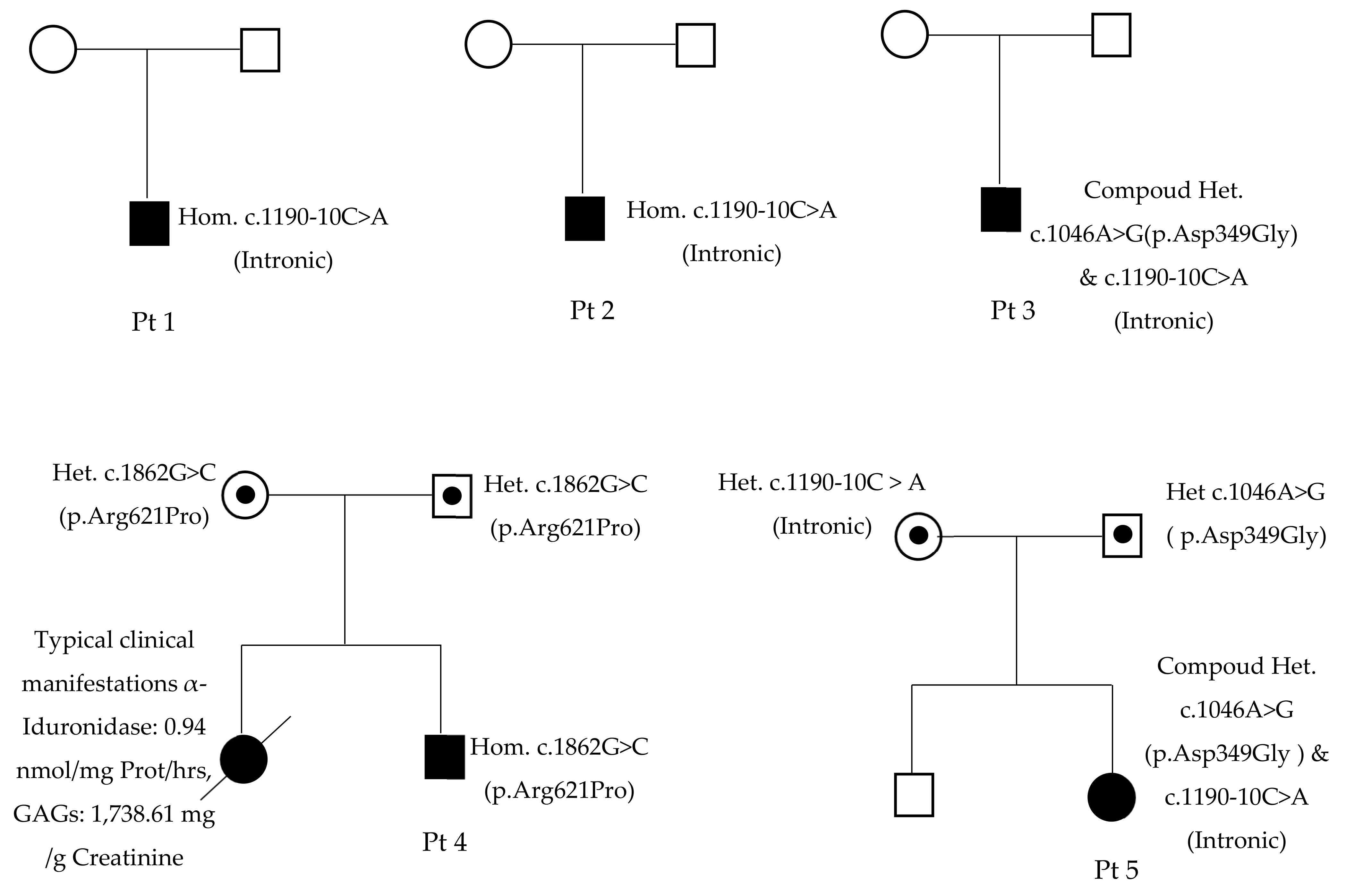
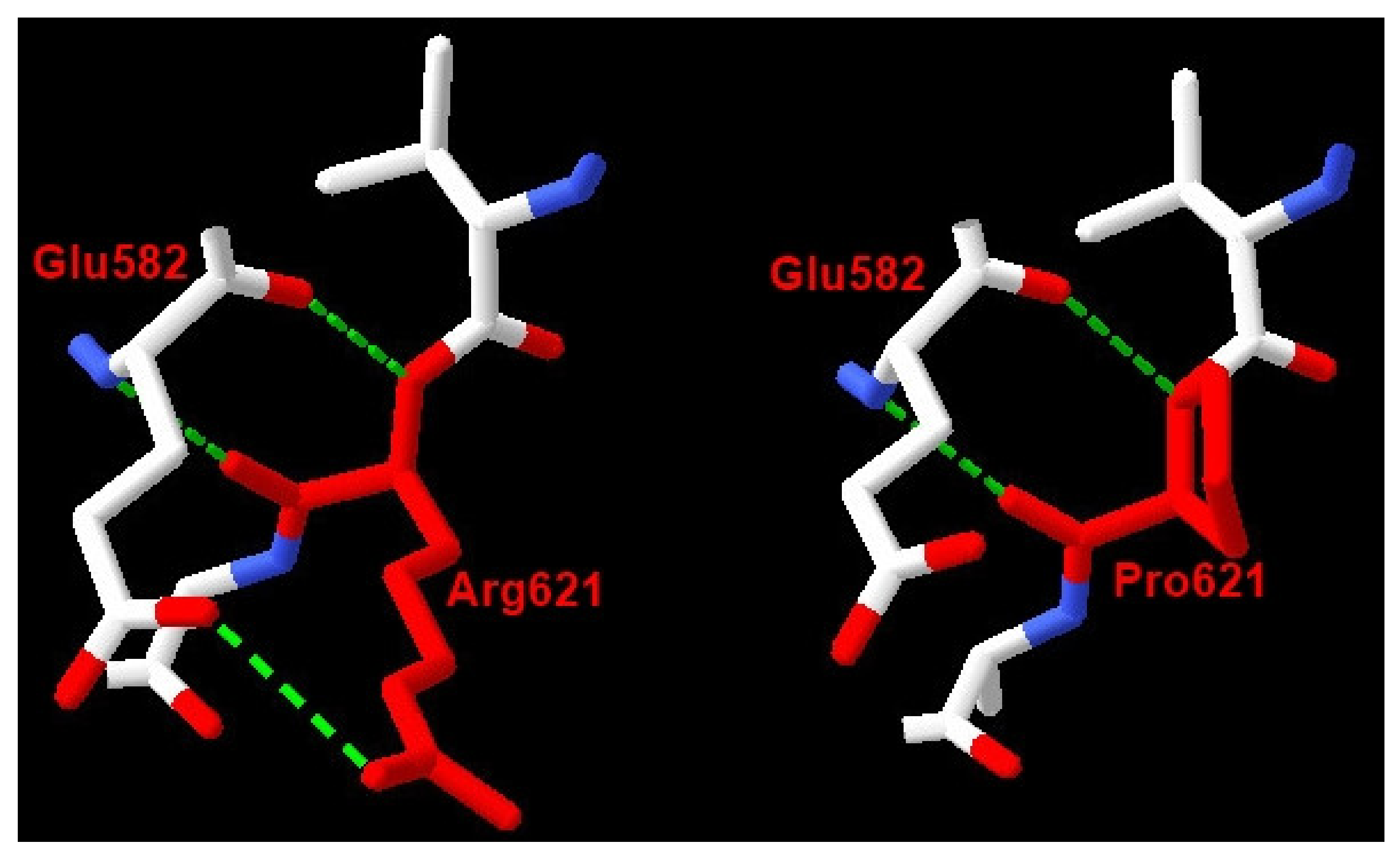
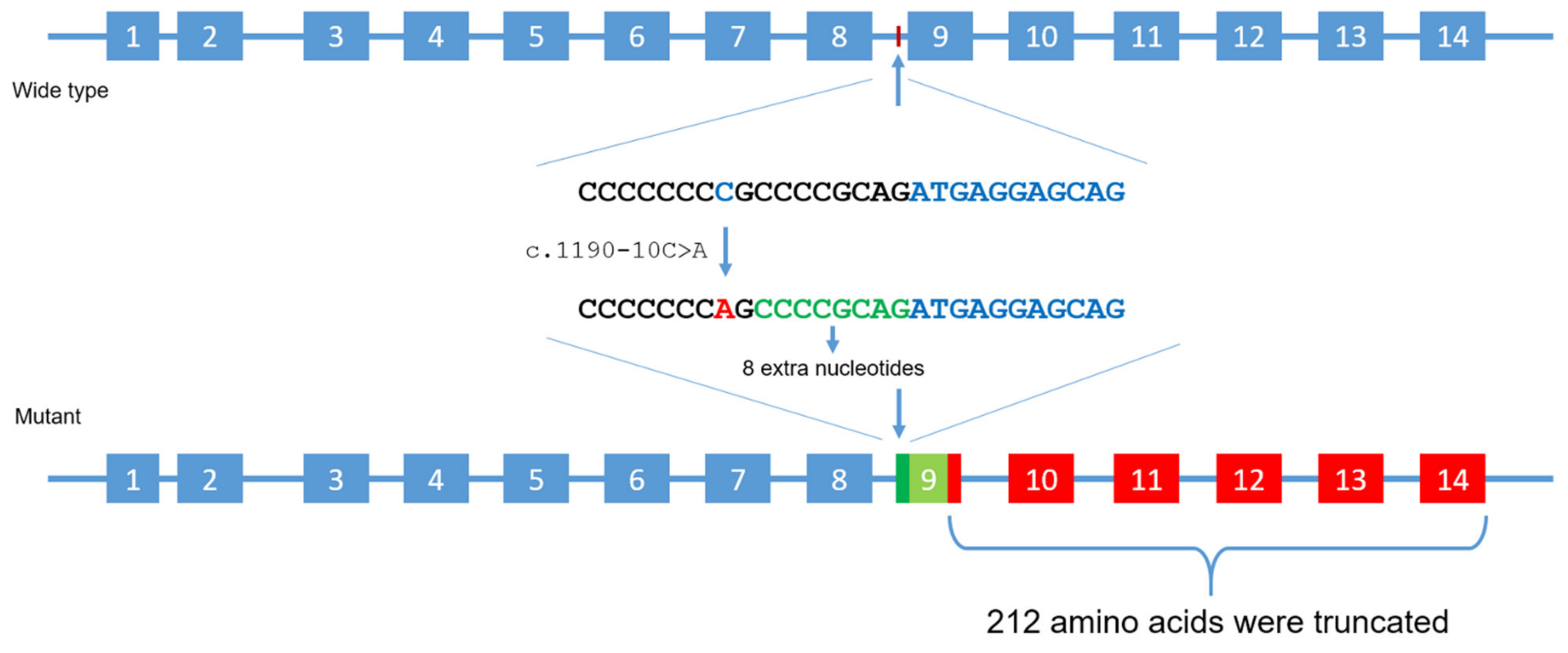
3.2. Molecular Genetic Analysis
4. Discussion
5. Conclusions
6. Limitation of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Vu, D.C.; Can, N.B.T.; Nguyen, K.N.; Bui, T.P.; Yoo, H.-W.; Kim, G.-H.; Hwu, W.-L. AB112. Spectrum of mucopolysaccharidoses in Vietnam. Ann. Transl. Med. 2017, 5, AB112. [Google Scholar] [CrossRef] [Green Version]
- Ngu, L.-H. Diagnosis and Management of Patients with Mucopolysaccharidoses in Malaysia. J. Mucopolysaccharidosis Rare Dis. 2018, 4, 11–13. [Google Scholar] [CrossRef]
- Cho, S.Y.; Sohn, Y.B.; Jin, D.-K. An overview of Korean patients with mucopolysaccharidosis and collaboration through the Asia Pacific MPS Network. Intractable Rare Dis. Res. 2014, 3, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atçeken, N.; Özgül, R.K.; Yilmaz, D.Y.; Tokatli, A.; Coşkun, T.; Sivri, H.S.; Dursun, A.; Karaca, M. Evaluation and identification of IDUA gene mutations in Turkish patients with mucopolysaccharidosis type I. Turk. J. Med. Sci. 2016, 46, 404–408. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM-Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 29 June 2021).
- Bertola, F.; Filocamo, M.; Casati, G.; Mort, M.; Rosano, C.; Tylki-Szymanska, A.; Tüysüz, B.; Gabrielli, O.; Grossi, S.; Scarpa, M.; et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: Identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum. Mutat. 2011, 32, E2189–E2210. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chuang, C.K.; Lin, S.P. Newborn Screening for Lysosomal Storage Diseases in Taiwan. J. Mucopolysaccharidosis Rare Dis. 2017, 3, 14–19. [Google Scholar] [CrossRef]
- Lim, K.H.; Fairbrother, W.G. Spliceman—A computational web server that predicts sequence variations in pre-mRNA splicing. Bioinformatics 2012, 28, 1031–1032. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Marín, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Mercer, J.; Mackinnon, S.; Yue, W.W.; Church, H.; E Beesley, C.; Broomfield, A.; Jones, S.; Tylee, K. IDUA mutational profile and genotype–phenotype relationships in UK patients with Mucopolysaccharidosis Type I. Hum. Mutat. 2017, 38, 1555–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, U.; Yogalingam, G.; Brooks, D.; Leistner-Segal, S.; Schwartz, I.V.D.; Lima, L.; Norato, D.Y.; Brum, J.M.; Beesley, C.; Winchester, B.; et al. Identification and characterization of 13 new mutations in mucopolysaccharidosis type I patients. Mol. Genet. Metab. 2003, 78, 37–43. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.; Clarke, L. Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [Green Version]
- Scott, H.; Bunge, S.; Gal, A.; Clarke, L.; Morris, P.; Hopwood, J.J. Molecular genetics of muccpolysaccharidosis type I: Diagnostic, clinical, and biological implications. Hum. Mutat. 1995, 6, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Venturi, N.; Rovelli, A.; Parini, R.; Menni, F.; Brambillasca, F.; Bertagnolio, F.; Uziel, G.; Gatti, R.; Filocamo, M.; Donati, M.; et al. Molecular analysis of 30 mucopolysaccharidosis type I patients: Evaluation of the mutational spectrum in Italian population and identification of 13 novel mutations. Hum. Mutat. 2002, 20, 231. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Phenotype prediction for mucopolysaccharidosis type I by in silico analysis. Orphanet. J. Rare Dis. 2017, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhabiles, H.; Jieshuang, J.; Lejeune, F. Nonsense Mutation Correction in Human Diseases: An Approach for Targeted Medicine; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef] [PubMed]
- Burset, M.; Seledtsov, I.; Solovyev, V. SpliceDB: Database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001, 29, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Uttarilli, A.; Ranganath, P.; Matta, D.; Jain, J.M.N.; Prasad, K.; Babu, A.; Girisha, K.; Verma, I.C.; Phadke, S.R.; Mandal, K.; et al. Identification and Characterization of 20 Novel Pathogenic Variants in 60 unrelated Indian patients with Mucopolysaccharidoses (MPS) type I and type II. Clin. Genet. 2016, 90, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wood, T.; Thompson, J.N. Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human α-l-iduronidase (IDUA) gene. Genet. Med. 2002, 4, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Pt1 | Pt2 | Pt3 | Pt4 | Pt5 |
|---|---|---|---|---|---|
| Age of diagnosis (months) | 20 | 15 | 38 | 9 | 54 |
| Age onset of the first symptoms (months) | 20 | 15 | 4 | 9 | 24 |
| The first symptoms | cornea clouding | coarse face | kyphosis | kyphosis | Joint stiffness |
| Coarse face | + | + | + | + | + |
| Cornea clouding | + | − | + | − | + |
| Joint stiffness | − | − | + | − | + |
| Long bone deformation | − | − | − | − | − |
| Spinal deformation | − | − | + | + | − |
| Hearing loss | − | + | NA | + | + |
| Valvular heart disease | + | + | + | − | − |
| Inguinal or umbilical hernia | − | − | + | + | − |
| Dermal melanocytosis | + | + | + | + | + |
| Hepatosplenomegaly | − | − | − | − | − |
| α-L-iduronidase (dried blood spots, normal >1.32 µM/h) | 0.17 | 0.06 | 0.13 | ||
| α-L-iduronidase (leukocytes, normal 15.9–41.8 nmol/mg prot/h) | 0.01 | 0.43 | |||
| Urine heparan sulfate (mg/mmol creatinine) | 8.26 (<0.67) | 55.44 (0.65–3.51) | 8.18 (<0.76) | ||
| Urine dermatan sulfate (mg/mmol creatinine) | 11.89 (<0.24) | 14.07 (0.14–0.90) | 11.55 (<0.21) | ||
| Urine keratan sulfate (mg/mmol creatinine) | 0.32 (< 0.73) | 2.08 (0.08–0.98) | 0.23 (<0.92) | ||
| Urine glycosaminoglycans (mg/g creatinine) | 1106.4 (10.7–112) | 508.8 (10.7–112) | |||
| Variant 1 | c.1190-10C>A | c.1190-10C>A | c.1190-10C>A | c.1862G>C | c.1190-10C>A |
| Variant 2 | c.1190-10C>A | c.1190-10C>A | c.1046A>G | c.1862G>C | c.1046A>G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Can, N.T.B.; Tran, D.M.; Bui, T.P.; Nguyen, K.N.; Nguyen, H.H.; Nguyen, T.V.; Hwu, W.-L.; Tomatsu, S.; Vu, D.C. Molecular Analysis of Vietnamese Patients with Mucopolysaccharidosis Type I. Life 2021, 11, 1162. https://doi.org/10.3390/life11111162
Can NTB, Tran DM, Bui TP, Nguyen KN, Nguyen HH, Nguyen TV, Hwu W-L, Tomatsu S, Vu DC. Molecular Analysis of Vietnamese Patients with Mucopolysaccharidosis Type I. Life. 2021; 11(11):1162. https://doi.org/10.3390/life11111162
Chicago/Turabian StyleCan, Ngoc Thi Bich, Dien Minh Tran, Thao Phuong Bui, Khanh Ngoc Nguyen, Hoang Huy Nguyen, Tung Van Nguyen, Wuh-Liang Hwu, Shunji Tomatsu, and Dung Chi Vu. 2021. "Molecular Analysis of Vietnamese Patients with Mucopolysaccharidosis Type I" Life 11, no. 11: 1162. https://doi.org/10.3390/life11111162
APA StyleCan, N. T. B., Tran, D. M., Bui, T. P., Nguyen, K. N., Nguyen, H. H., Nguyen, T. V., Hwu, W.-L., Tomatsu, S., & Vu, D. C. (2021). Molecular Analysis of Vietnamese Patients with Mucopolysaccharidosis Type I. Life, 11(11), 1162. https://doi.org/10.3390/life11111162