
Initial Screening of Poly(ethylene glycol) Amino Ligands for Affinity Purification of Plasmid DNA in Aqueous Two-Phase Systems

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
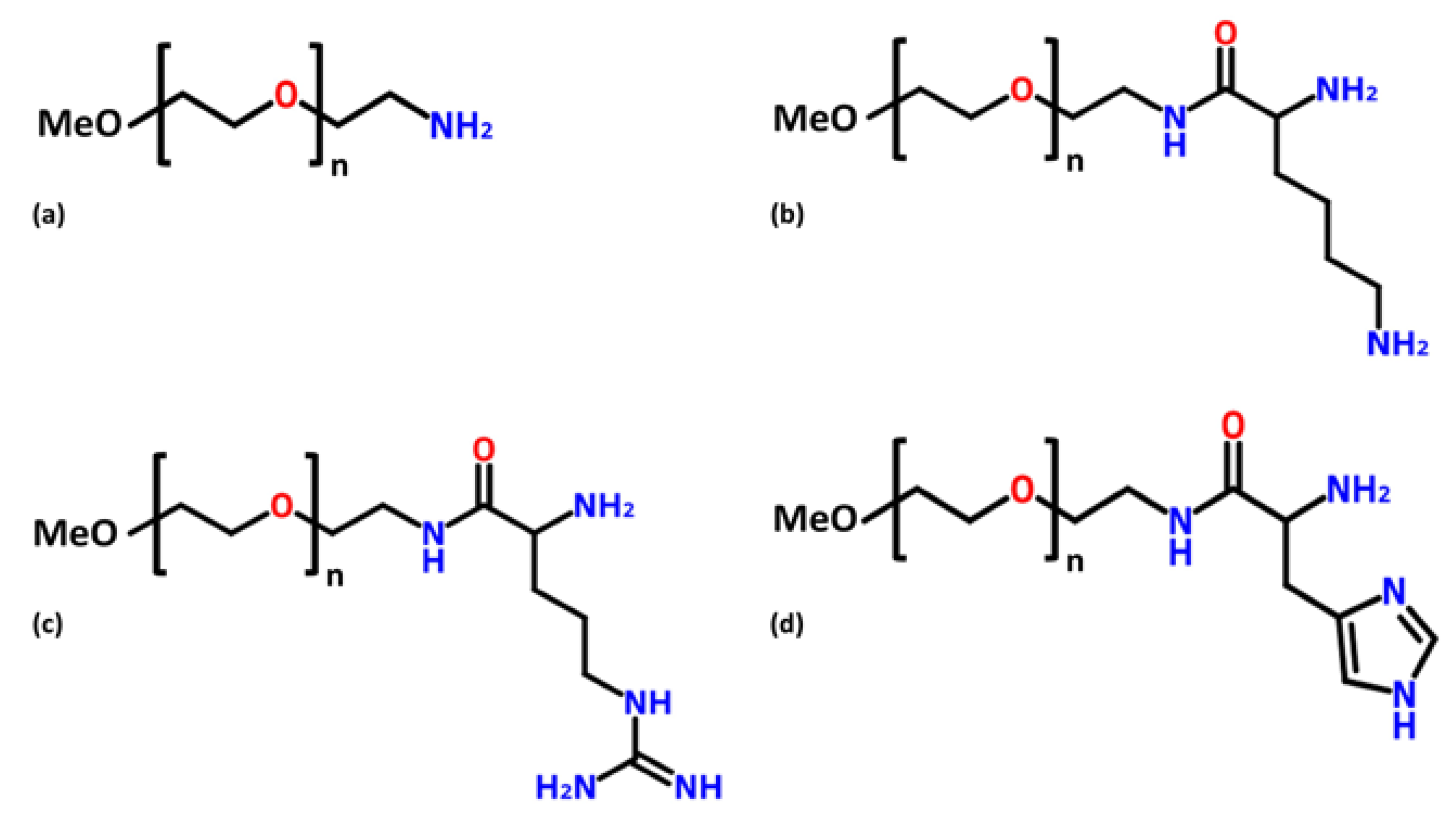
2.2. Poly(Ethylene Glycol)–Amine Affinity Ligands Synthesis
2.2.1. Methoxypolyethyleneglycol–Lysine and –Arginine
2.2.2. Methoxypolyethyleneglycol–Histidine
2.3. Plasmid Production
2.4. Alkaline Cell Lysis
2.5. Lysate Desalting
2.6. Aqueous Two-Phase Experiments
2.7. Extraction with an Ammonium-Sulphate-Rich Phase
2.8. Nucleic Acids Partitioning Analysis by Agarose Gel Electrophoresis (AGE)
2.9. Total Protein Partitioning Analysis Using Polyacrylamide Denaturing Gel Electrophoresis (SDS–PAGE)
2.10. Total Protein Quantification
2.11. Densitometric Analysis of Agarose Gel Bands
3. Results and Discussion
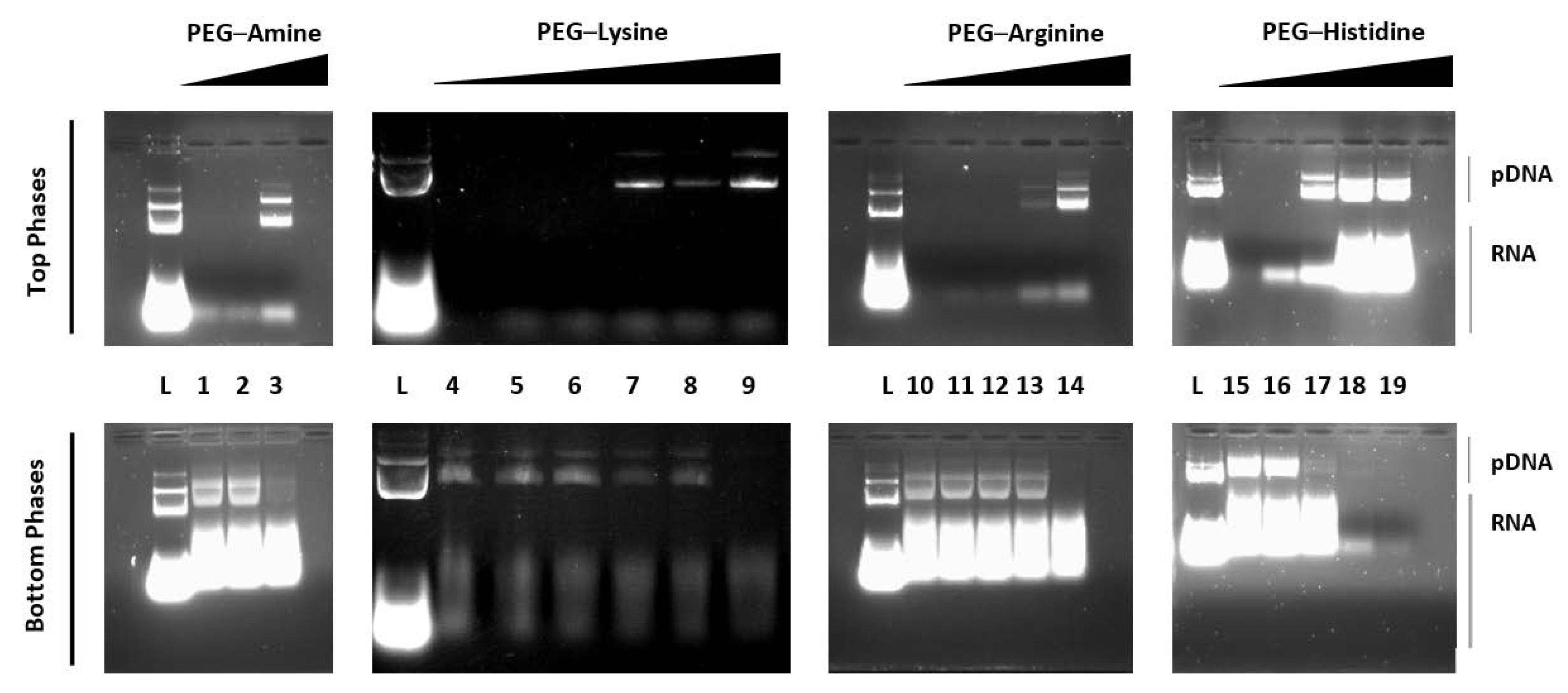
3.1. Specificity of PEG–Amino Affinity Ligands
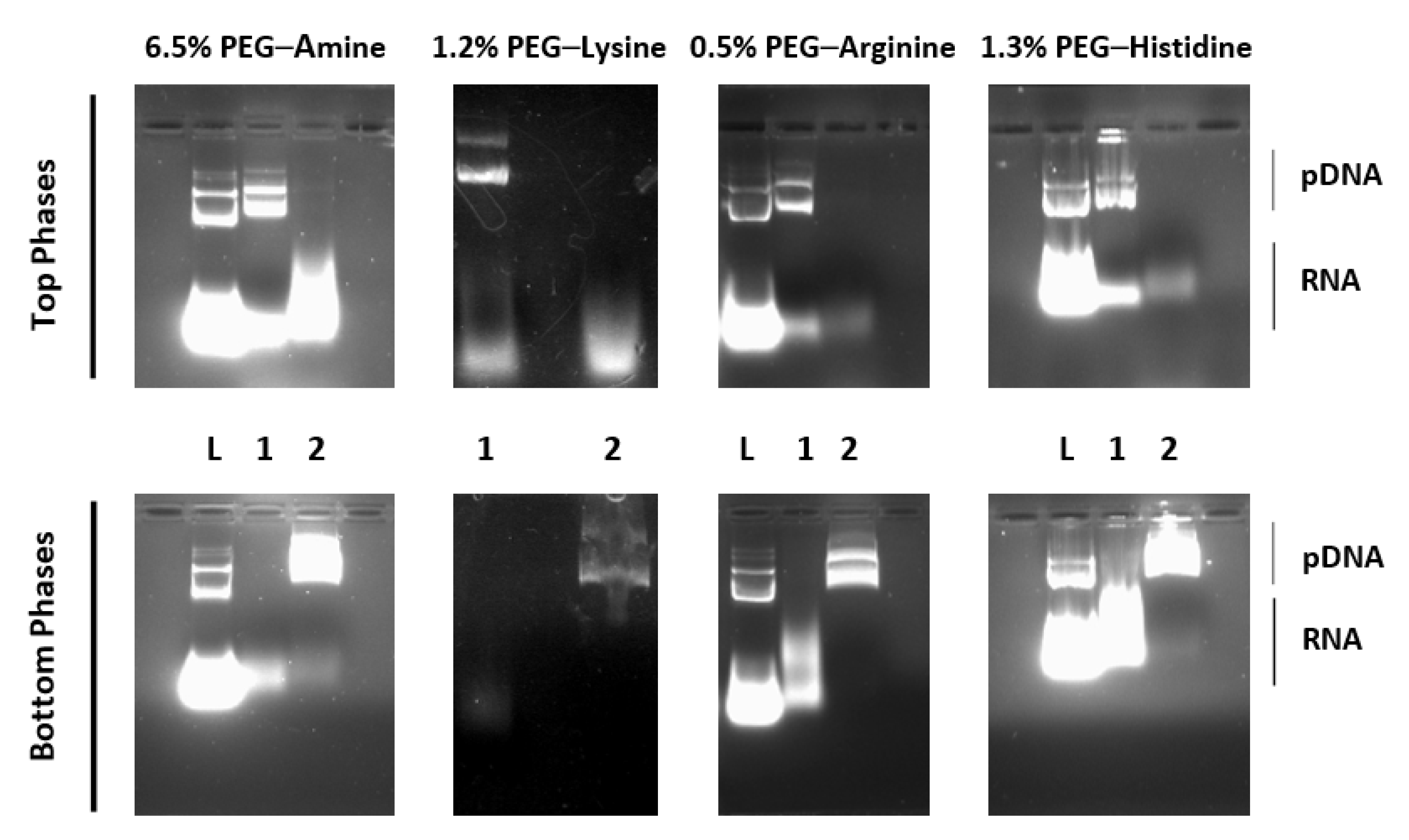
3.2. pDNA Extraction with an Ammonium-Sulphate-Rich Phase
3.3. Protein Contamination Assessment
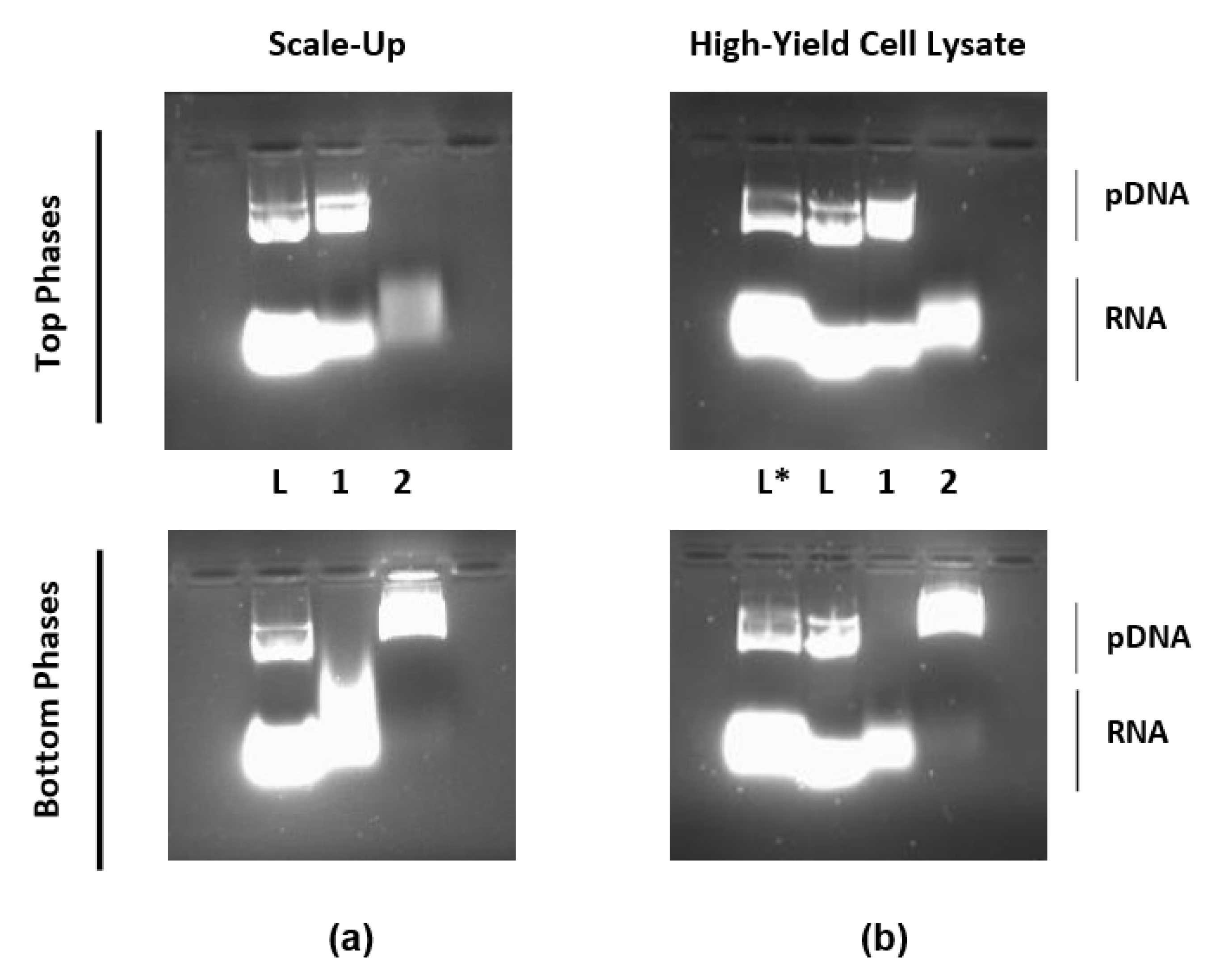
3.4. Process Scale-Up and Capacity Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Valente, J.; Queiroz, J.; Sousa, F. Dilemma on plasmid DNA purification: Binding capacity vs selectivity. J. Chromatogr. A 2021, 1637, 461848. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Kim, S.W. Recent Advances in the Development of Bio-Reducible Polymers for Efficient Cancer Gene Delivery Systems. Cancer Med. J. 2019, 2, 6–13. [Google Scholar]
- Ponti, F.; Campolungo, M.; Melchiori, C.; Bono, N.; Candiani, G. Cationic lipids for gene delivery: Many players, one goal. Chem. Phys. Lipids 2021, 235, 105032. [Google Scholar] [CrossRef]
- Tejeda-Mansir, A.; Rendon, A.G.; Guerrero-German, P. Plasmid-DNA lipid and polymeric nanovaccines: A new strategic in vaccines development. Biotechnol. Genet. Eng. Rev. 2019, 35, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.M.; Moreira, G.M.S.G.; Mendonça, M. DNA vaccines against COVID-19: Perspectives and challenges. Life Sci. 2021, 267, 118919. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mohammadi-Samani, S.; Firouzabadi, N.; Dehshahri, A.; Vazin, A. A focused review on technologies, mechanisms, safety, and efficacy of available COVID-19 vaccines. Int. Immunopharmacol. 2021, 100, 108162. [Google Scholar] [CrossRef]
- Mallapaty, S. India’s DNA COVID vaccine is a world first—More are coming. Nat. Cell Biol. 2021, 597, 161–162. [Google Scholar] [CrossRef]
- Zieneldien, T.; Kim, J.; Cao, J.; Cao, C. COVID-19 Vaccines: Current Conditions and Future Prospects. Biology 2021, 10, 960. [Google Scholar] [CrossRef]
- Abdulrahman, A.; Ghanem, A. Recent advances in chromatographic purification of plasmid DNA for gene therapy and DNA vaccines: A review. Anal. Chim. Acta 2018, 1025, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Healey, R.; Adly, F.G. Current trends in separation of plasmid DNA vaccines: A review. Anal. Chim. Acta 2013, 760, 1–15. [Google Scholar] [CrossRef]
- Shamlou, P.A. Scaleable processes for the manufacture of therapeutic quantities of plasmid DNA. Biotechnol. Appl. Biochem. 2003, 37, 207–218. [Google Scholar] [CrossRef]
- Diogo, M.M.; Queiroz, J.; Prazeres, D. Chromatography of plasmid DNA. J. Chromatogr. A 2005, 1069, 3–22. [Google Scholar] [CrossRef]
- Urthaler, J.; Schuchnigg, H.; Garidel, P.; Huber, H. Industrial Manufacturing of Plasmid-DNA Products for Gene Vaccination and Therapy. In Gene Vaccines; Thalhamer, J., Weiss, R., Scheiblhofer, S., Eds.; Springer: Vienna, Austria, 2012; pp. 311–330. ISBN 978-3-7091-0438-5, 978-3-7091-0439-2. [Google Scholar]
- Phong, W.N.; Show, P.L.; Chow, Y.H.; Ling, T.C. Recovery of biotechnological products using aqueous two phase systems. J. Biosci. Bioeng. 2018, 126, 273–281. [Google Scholar] [CrossRef]
- Khoo, K.S.; Leong, H.Y.; Chew, K.W.; Lim, J.-W.; Ling, T.C.; Show, P.L.; Yen, H.-W. Liquid Biphasic System: A Recent Bioseparation Technology. Processes 2020, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.F.B.; Coutinho, J.A.P. Chapter 5—Aqueous Two-Phase Systems. In Liquid-Phase Extraction; Handbooks in Separation Science; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 157–182. ISBN 978-0-12-816911-7. [Google Scholar]
- Zaslavsky, B.Y. Aqueous Two-Phase Partitioning: Physical Chemistry and Bioanalytical Applications; Marcel Dekker: New York, NY, USA, 1995. [Google Scholar]
- González-Valdez, J.; Mayolo-Deloisa, K.; Rito-Palomares, M. Novel aspects and future trends in the use of aqueous two-phase systems as a bioengineering tool. J. Chem. Technol. Biotechnol. 2018, 93, 1836–1844. [Google Scholar] [CrossRef]
- Torres-Acosta, M.A.; Mayolo-Deloisa, K.; González-Valdez, J.; Rito-Palomares, M. Aqueous Two-Phase Systems at Large Scale: Challenges and Opportunities. Biotechnol. J. 2019, 14, e1800117. [Google Scholar] [CrossRef] [Green Version]
- Kaplanow, I.; Goerzgen, F.; Merz, J.; Schembecker, G. Mass Transfer of Proteins in Aqueous Two-Phase Systems. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ruiz, F.; Benavides, J.; Rito-Palomares, M. Affinity ATPS Strategies for the Selective Fractionation of Biomolecules. In Aqueous Two-Phase Systems for Bioprocess Development for the Recovery of Biological Products; Rito-Palomares, M., Benavides, J., Eds.; Food Engineering Series; Springer International Publishing: Cham, Switzerland, 2017; pp. 97–121. ISBN 978-3-319-59309-8. [Google Scholar]
- Silva, M.F.; Fernandes-Platzgummer, A.; Aires-Barros, M.R.; Azevedo, A.M. Integrated purification of monoclonal antibodies directly from cell culture medium with aqueous two-phase systems. Sep. Purif. Technol. 2014, 132, 330–335. [Google Scholar] [CrossRef]
- Boland, M.J. Aqueous Two-Phase Extraction and Purification of Animal Proteins. Mol. Biotechnol. 2002, 20, 085–094. [Google Scholar] [CrossRef]
- Show, P.-L.; Ling, T.-C.; Lan, J.C.-W.; Tey, B.-T.; Ramanan, R.N.; Yong, S.-T.; Ooi, C.-W. Review of Microbial Lipase Purifica-tion Using Aqueous Two-Phase Systems. Curr. Pharm. Design 2015, 19, 19–29. [Google Scholar]
- Platis, D.; Labrou, N. Development of an aqueous two-phase partitioning system for fractionating therapeutic proteins from tobacco extract. J. Chromatogr. A 2006, 1128, 114–124. [Google Scholar] [CrossRef]
- Azevedo, A.M.; Rosa, P.; Ferreira, I.; Pisco, A.O.; de Vries, J.; Korporaal, R.; Visser, T.; Aires-Barros, M. Affinity-enhanced purification of human antibodies by aqueous two-phase extraction. Sep. Purif. Technol. 2009, 65, 31–39. [Google Scholar] [CrossRef]
- Yücekan, I.; Önal, S. Partitioning of invertase from tomato in poly(ethylene glycol)/sodium sulfate aqueous two-phase systems. Process. Biochem. 2011, 46, 226–232. [Google Scholar] [CrossRef]
- Maestro, B.; Velasco, I.; Castillejo, I.; Arévalo-Rodríguez, M.; Cebolla, Á.; Sanz, J.M. Affinity partitioning of proteins tagged with choline-binding modules in aqueous two-phase systems. J. Chromatogr. A 2008, 1208, 189–196. [Google Scholar] [CrossRef]
- Roque, A.C.; Silva, C.S.; Taipa, M. Ângela Affinity-based methodologies and ligands for antibody purification: Advances and perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Show, P.L.; Tan, C.P.; Anuar, M.S.; Ariff, A.; Yusof, Y.A.; Chen, S.K.; Ling, T.C. Extractive fermentation for improved production and recovery of lipase derived from Burkholderia cepacia using a thermoseparating polymer in aqueous two-phase systems. Bioresour. Technol. 2012, 116, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, A.M.; Rosa, P.A.; Ferreira, I.F.; Aires-Barros, M.R. Chromatography-free recovery of biopharmaceuticals through aqueous two-phase processing. Trends Biotechnol. 2009, 27, 240–247. [Google Scholar] [CrossRef]
- Asenjo, J.A.; Andrews, B.A. Aqueous two-phase systems for protein separation: A perspective. J. Chromatogr. A 2011, 1218, 8826–8835. [Google Scholar] [CrossRef]
- Nadar, S.; Pawar, R.G.; Rathod, V.K. Recent advances in enzyme extraction strategies: A comprehensive review. Int. J. Biol. Macromol. 2017, 101, 931–957. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, M.; Wang, X.; Wu, S.; Li, D.; Liu, C.; Feng, Z.; Li, J. Effective separation of prolyl endopeptidase from Aspergillus Niger by aqueous two phase system and its characterization and application. Int. J. Biol. Macromol. 2021, 169, 384–395. [Google Scholar] [CrossRef]
- Khan, B.M.; Cheong, K.-L.; Liu, Y. ATPS: “Aqueous two-phase system” as the “answer to protein separation” for protein-processing food industry. Crit. Rev. Food Sci. Nutr. 2018, 59, 3165–3178. [Google Scholar] [CrossRef] [PubMed]
- Varadavenkatesan, T.; Pai, S.; Vinayagam, R.; Pugazhendhi, A.; Selvaraj, R. Recovery of value-added products from wastewater using Aqueous Two-Phase Systems—A review. Sci. Total. Environ. 2021, 778, 146293. [Google Scholar] [CrossRef]
- Ohlsson, R.; Hentschel, C.C.; Williams, J.G. A rapid method for the isolation of circular DNA using an aqueous two-phase partition system. Nucleic Acids Res. 1978, 5, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, S.C.; Monteiro, G.; Cabral, J.M.; Prazeres, D.M.F. Isolation of plasmid DNA from cell lysates by aqueous two-phase systems. Biotechnol. Bioeng. 2002, 78, 376–384. [Google Scholar] [CrossRef]
- Trindade, I.P.; Diogo, M.M.; Prazeres, D.M.; Marcos, J.C. Purification of plasmid DNA vectors by aqueous two-phase extraction and hydrophobic interaction chromatography. J. Chromatogr. A 2005, 1082, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.; Azevedo, A.M.; Aires-Barros, M.R.; Prazeres, D.M.F. Purification of plasmid DNA with aqueous two phase systems of PEG 600 and sodium citrate/ammonium sulfate. Sep. Purif. Technol. 2009, 65, 22–30. [Google Scholar] [CrossRef]
- Wiendahl, M.; Oelmeier, S.A.; Dismer, F.; Hubbuch, J. High-throughput screening-based selection and scale-up of aqueous two-phase systems for pDNA purification. J. Sep. Sci. 2012, 35, 3197–3207. [Google Scholar] [CrossRef]
- Müller, M.; Frerix, A.; Kula, M.-R.; Hubbuch, J. Scalable recovery of plasmid DNA based on aqueous two-phase separation. Biotechnol. Appl. Biochem. 2005, 42, 57–66. [Google Scholar] [CrossRef]
- Kepka, C.; Collet, E.; Persson, J.; Ståhl, Å.; Lagerstedt, T.; Tjerneld, F.; Veide, A. Pilot-scale extraction of an intracellular recombinant cutinase from E. coli cell homogenate using a thermoseparating aqueous two-phase system. J. Biotechnol. 2003, 103, 165–181. [Google Scholar] [CrossRef]
- Duarte, S.P.; Fortes, A.G.; Prazeres, D.M.; Marcos, J.C. Preparation of plasmid DNA polyplexes from alkaline lysates by a two-step aqueous two-phase extraction process. J. Chromatogr. A 2007, 1164, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, H.; Hine, A.; Brocchini, S.; Slater, N.; Marcos, J. Affinity partitioning of plasmid DNA with a zinc finger protein. J. Chromatogr. A 2008, 1206, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, H.; Hine, A.; Brocchini, S.; Slater, N.; Marcos, J.C. Dual affinity method for plasmid DNA purification in aqueous two-phase systems. J. Chromatogr. A 2010, 1217, 1429–1436. [Google Scholar] [CrossRef]
- Nazer, B.; Dehghani, M.R.; Goliaei, B. Plasmid DNA affinity partitioning using polyethylene glycol – sodium sulfate aqueous two-phase systems. J. Chromatogr. B 2017, 1044-1045, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.M.; Laskowski, R.A.; Thornton, J.M. Amino Acid–Base Interactions: A Three-Dimensional Analysis of Protein–DNA Interactions at an Atomic Level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, M.M. AANT: The Amino Acid-Nucleotide Interaction Database. Nucleic Acids Res. 2004, 32, 174D–181D. [Google Scholar] [CrossRef] [Green Version]
- Sousa, F.; Cruz, C.; Queiroz, J.A. Amino acids-nucleotides biomolecular recognition: From biological occurrence to affinity chromatography. J. Mol. Recognit. 2010, 23, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Sousa, Â.; Sousa, F.; Queiroz, J. Advances in chromatographic supports for pharmaceutical-grade plasmid DNA purification: Liquid Chromatography. J. Sep. Sci. 2012, 35, 3046–3058. [Google Scholar] [CrossRef]
- Sousa, A.; Sousa, F.; Queiroz, J. Impact of lysine-affinity chromatography on supercoiled plasmid DNA purification. J. Chromatogr. B 2011, 879, 3507–3515. [Google Scholar] [CrossRef] [PubMed]
- Sousa, Â.; Sousa, F.; Queiroz, J.A. Differential interactions of plasmid DNA, RNA and genomic DNA with amino acid-based affinity matrices. J. Sep. Sci. 2010, 33, 2610–2618. [Google Scholar] [CrossRef]
- Sousa, F. A importância da purificação de plasmídeos para terapia génica. Boletim Sociedade Portuguesa de Biotecnologia: Biotecnologia na Saúde; Sociedade Portuguesa de Biotecnologia: Braga, Portugal, 2013; Volume 2, pp. 24–27. [Google Scholar]
- Freitas, S.; Sousa, F.; Azzoni, A.R.; Prazeres, D.M.F.; Queiroz, J. Selective purification of supercoiled plasmid DNA from clarified cell lysates with a single histidine–agarose chromatography step. Biotechnol. Appl. Biochem. 2006, 45, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Prazeres, D.; Queiroz, J. Circular dichroism investigation of the effect of plasmid DNA structure on retention in histidine chromatography. Arch. Biochem. Biophys. 2007, 467, 154–162. [Google Scholar] [CrossRef]
- Sousa, F.; Prazeres, D.M.; Queiroz, J. Improvement of transfection efficiency by using supercoiled plasmid DNA purified with arginine affinity chromatography. J. Gene Med. 2008, 11, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Matos, T.; Prazeres, D.; Queiroz, J. Specific recognition of supercoiled plasmid DNA in arginine affinity chromatography. Anal. Biochem. 2008, 374, 432–434. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning. A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef]
- Heukeshoven, J.; Dernick, R. Simplified method for silver staining of proteins in polyacrylamide gels and the mechanism of silver staining. Electrophoresis 1985, 6, 103–112. [Google Scholar] [CrossRef]
- O’Connell, K.L.; Stults, J.T. Identification of mouse liver proteins on two-dimensional electrophoresis gels by matrix-assisted laser desorption/ionization mass spectrometry ofin situ enzymatic digests. Electrophoresis 1997, 18, 349–359. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensistive Method for Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. 2011. Available online: https://imagej.nih.gov/ij/ (accessed on 15 September 2021).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- da Silva, N.; Ferreira, L.A.; Madeira, P.; Teixeira, J.; Uversky, V.N.; Zaslavsky, B.Y. Effect of sodium chloride on solute–solvent interactions in aqueous polyethylene glycol–sodium sulfate two-phase systems. J. Chromatogr. A 2015, 1425, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepka, C.; Rhodin, J.; Lemmens, R.; Tjerneld, F.; Gustavsson, P.-E. Extraction of plasmid DNA from Escherichia coli cell lysate in a thermoseparating aqueous two-phase system. J. Chromatogr. A 2004, 1024, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Tomaz, C.; Prazeres, D.; Queiroz, J. Separation of supercoiled and open circular plasmid DNA isoforms by chromatography with a histidine–agarose support. Anal. Biochem. 2005, 343, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Queiroz, J. Supercoiled plasmid quality assessment by analytical arginine-affinity chromatography. J. Chromatogr. A 2010, 1218, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, H. Theory of phase formation in aqueous two-phase systems. J. Chromatogr. B Biomed. Sci. Appl. 1996, 680, 3–30. [Google Scholar] [CrossRef]
- Barbosa, H.; Slater, N.K.; Marcos, J.C. Protein quantification in the presence of poly(ethylene glycol) and dextran using the Bradford method. Anal. Biochem. 2009, 395, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 1–22. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6.5% PEG–Amine | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 4525.3 | 5.1 | 58,908.5 | 74.1 |
| RNA | 84,927.4 | 94.9 | 20,622.5 | 25.9 |
| 1.2% PEG–Lysine | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 0 | 0 | 50,694.5 | 100 |
| RNA | 43254.3 | 100 | 0 | 0 |
| 0.5% PEG–Arginine | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 0 | 0 | 48,787.4 | 100 |
| RNA | 54,061.7 | 100 | 0 | 0 |
| 1.3% PEG–Histidine | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 0 | 0 | 47,470.7 | 81.3 |
| RNA | 30,013.5 | 100 | 10,904.5 | 18.7 |
| 0.5% PEG–Arginine (Scale-Up) | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 0 | 0 | 29,553.5 | 77.5 |
| RNA | 39,394.4 | 100 | 8601.0 | 22.5 |
| 0.5% PEG–Arginine (High-Yield Cell Lysate) | ||||
| Top Phase | Bottom Phase | |||
| Band Area | Percentage | Band Area | Percentage | |
| pDNA | 0 | 0 | 24,504.4 | 81.8 |
| RNA | 28,249.9 | 100 | 5453.5 | 18.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, N.R.; Jorge, P.; Martins, J.A.; Teixeira, J.A.; Marcos, J.C. Initial Screening of Poly(ethylene glycol) Amino Ligands for Affinity Purification of Plasmid DNA in Aqueous Two-Phase Systems. Life 2021, 11, 1138. https://doi.org/10.3390/life11111138
da Silva NR, Jorge P, Martins JA, Teixeira JA, Marcos JC. Initial Screening of Poly(ethylene glycol) Amino Ligands for Affinity Purification of Plasmid DNA in Aqueous Two-Phase Systems. Life. 2021; 11(11):1138. https://doi.org/10.3390/life11111138
Chicago/Turabian Styleda Silva, Nuno R., Paula Jorge, José A. Martins, José A. Teixeira, and João C. Marcos. 2021. "Initial Screening of Poly(ethylene glycol) Amino Ligands for Affinity Purification of Plasmid DNA in Aqueous Two-Phase Systems" Life 11, no. 11: 1138. https://doi.org/10.3390/life11111138
APA Styleda Silva, N. R., Jorge, P., Martins, J. A., Teixeira, J. A., & Marcos, J. C. (2021). Initial Screening of Poly(ethylene glycol) Amino Ligands for Affinity Purification of Plasmid DNA in Aqueous Two-Phase Systems. Life, 11(11), 1138. https://doi.org/10.3390/life11111138