
NMUR1 in the NMU-Mediated Regulation of Bone Remodeling




Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Gene Expression Studies
2.3. Antibody Array
2.4. Mice
2.5. Micro Computed Tomography (µCT)
2.6. Histological Analyses
2.7. Serum Analysis
2.8. Statistical Analyses
3. Results
3.1. Signaling Changes in NMU Knockout Bones
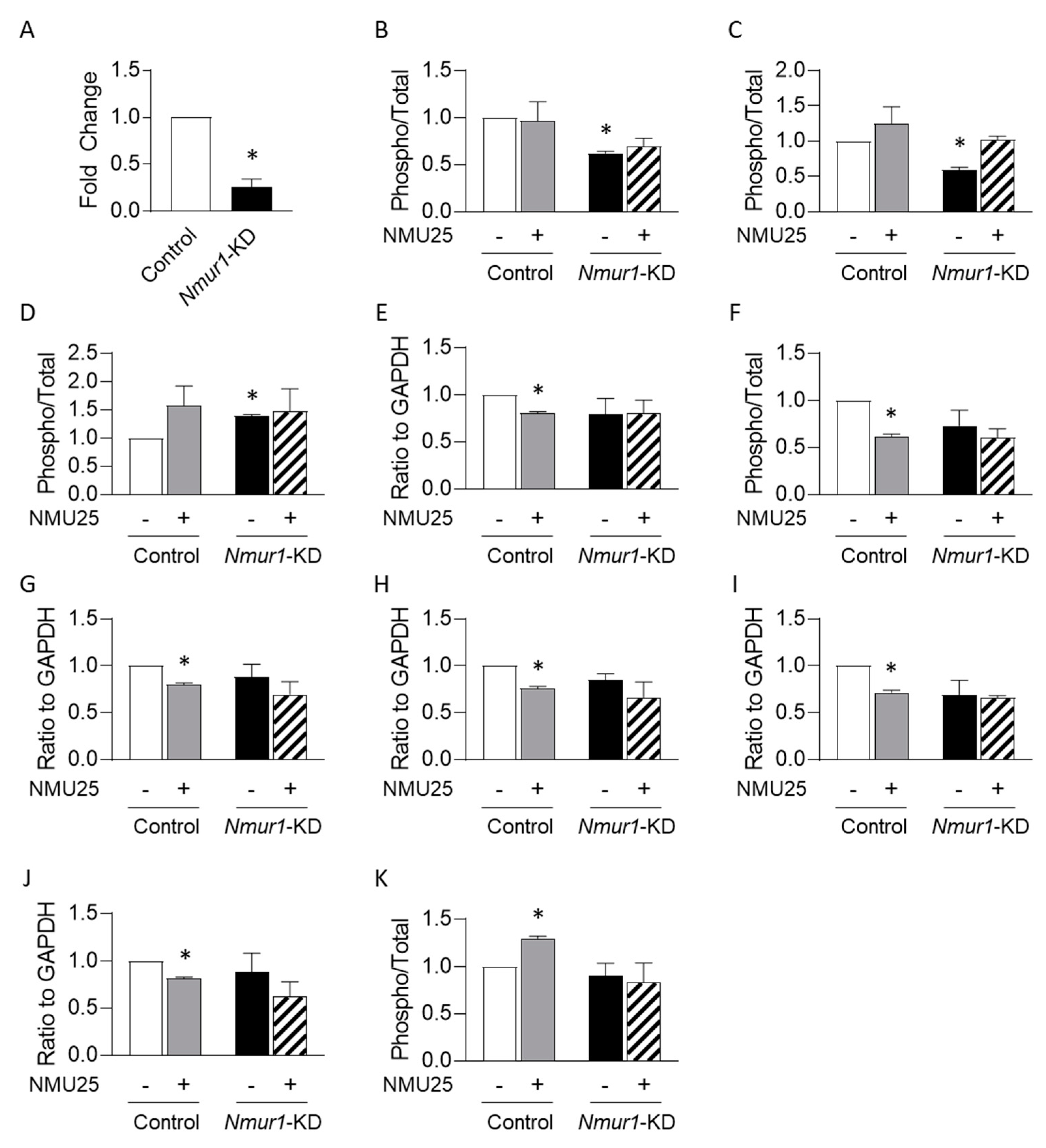
3.2. NMU Regulates Osteoblast Function through NMUR1 In Vitro
3.3. Bone Volume Is Unchanged in the Absence of NMUR1
3.4. Histological and Serum Analyses of NMUR1-Deficient Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leboime, A.; Confavreux, C.B.; Mehsen, N.; Paccou, J.; David, C.; Roux, C. Osteoporosis and mortality. Jt. Bone Spine 2010, 77 (Suppl. S2), S107–S112. [Google Scholar] [CrossRef]
- Singer, A.; Exuzides, A.; Spangler, L.; O’Malley, C.; Colby, C.; Johnston, K.; Agodoa, I.; Baker, J.; Kagan, R. Burden of Illness for Osteoporotic Fractures Compared With Other Serious Diseases Among Postmenopausal Women in the United States. Mayo Clin. Proc. 2015, 90, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Camacho, P.M.; Petak, S.M.; Binkley, N.; Clarke, B.L.; Harris, S.T.; Hurley, D.L.; Kleerekoper, M.; Lewiecki, E.M.; Miller, P.D.; Narula, H.S.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology Clinical Practice Guidelines for the Diagnosis and Treatment of Postmenopausal Osteoporosis—2016. Endocr. Pract. 2016, 22 (Suppl. S4), 1–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gullberg, B.; Johnell, O.; Kanis, J. World-wide Projections for Hip Fracture. Osteoporos. Int. 1997, 7, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Brighton, P.J.; Szekeres, P.G.; Willars, G.B. Neuromedin U and Its Receptors: Structure, Function, and Physiological Roles. Pharmacol. Rev. 2004, 56, 231–248. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.M.; Auger, J.L.; Gaillard, P.; Weissleder, R.; Wada, E.; Torres, R.; Kojima, M.; Benoist, C.; Mathis, D.; Binstadt, B.A. The neuropeptide neuromedin U promotes autoantibody-mediated arthritis. Arthritis Res. Ther. 2012, 14, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.-E.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hanada, R.; Kimura, A.; Abe, T.; Matsumoto, T.; Iwasaki, M.; Inose, H.; Ida, T.; Mieda, M.; Takeuchi, Y.; et al. Central control of bone remodeling by neuromedin U. Nat. Med. 2007, 13, 1234–1240. [Google Scholar] [CrossRef]
- Hsiao, Y.-T.; Jestes, K.J.; Jackson, K.L.; Zukosky, T.; Squire, M.E.; Hum, J.M.; Lowery, J.W. Neuromedin U (NMU) regulates osteoblast differentiation and activity. Biochem. Biophys. Res. Commun. 2020, 524, 890–894. [Google Scholar] [CrossRef]
- Zeng, H.; Gragerov, A.; Hohmann, J.G.; Pavlova, M.N.; Schimpf, B.A.; Xu, H.; Wu, L.-J.; Toyoda, H.; Zhao, M.-G.; Rohde, A.D.; et al. Neuromedin U Receptor 2-Deficient Mice Display Differential Responses in Sensory Perception, Stress, and Feeding. Mol. Cell. Biol. 2006, 26, 9352–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malendowicz, L.K.; Ziolkowska, A.; Rucinski, M. Neuromedins U and S involvement in the regulation of the hypothalamo–pituitary–adrenal axis. Front. Endocrinol. 2012, 3, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Mahlakoiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.-L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nat. Cell Biol. 2017, 549, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Eaton, M.S.; Weinstein, N.; Newby, J.B.; Plattes, M.M.; Foster, H.E.; Arthur, J.W.; Ward, T.D.; Shively, S.R.; Shor, R.; Nathan, J.; et al. Loss of the nutrient sensor TAS1R3 leads to reduced bone resorption. J. Physiol. Biochem. 2017, 74, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Dempster, D.W.; Compston, J.E.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2012, 28, 2–17. [Google Scholar] [CrossRef] [Green Version]
- Rucinski, M.; Ziolkowska, A.; Tyczewska, M.; Szyszka, M.; Malendowicz, L.K. Neuromedin U directly stimulates growth of cultured rat calvarial osteoblast-like cells acting via the NMU receptor 2 isoform. Int. J. Mol. Med. 2008, 22, 363–368. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Parameter | Wild Type | Nmur1 Knockout | p Value | |
|---|---|---|---|---|
| Distal Femora | TV (mm3) | 4.639 ± 0.256 | 4.567 ± 0.108 | 0.778 |
| BV/TV (ratio) | 0.154 ± 0.023 | 0.195 ± 0.026 | 0.289 | |
| Tb.N (#/mm) | 4.767 ±.1956 | 5.387 ± 0.317 | 0.165 | |
| Tb.Th (mm) | 0.049 ± 0.003 | 0.050 ± 0.001 | 0.893 | |
| Tb.Sp (mm) | 0.204 ± 0.009 | 0.179 ± 0.011 | 0.145 | |
| Conn.D (#/mm3) | 161.456 ± 18.937 | 228.086 ± 32.950 | 0.148 | |
| SMI | 2.201 ± 0.235 | 1.828 ± 0.205 | 0.262 | |
| Proximal Tibiae | TV (mm3) | 1.580 ± 0.099 | 1.752 ± 0.078 | 0.198 |
| BV/TV (ratio) | 0.195 ± 0.024 | 0.230 ± 0.021 | 0.314 | |
| Tb.N (#/mm) | 5.554 ± 0.161 | 6.051 ± 0.253 | 0.165 | |
| Tb.Th (mm) | 0.051 ± 0.002 | 0.053 ± 0.001 | 0.585 | |
| Tb.Sp (mm) | 0.168 ± 0.007 | 0.152 ± 0.007 | 0.169 | |
| Conn.D (#/mm3) | 176.637 ± 21.492 | 235.875 ± 22.430 | 0.096 | |
| SMI | 2.091 ± 0.198 | 1.873 ± 0.151 | 0.394 |
| Parameter | Wild Type | Nmur1 Knockout | p Value | |
|---|---|---|---|---|
| Mid-diaphysis Femora | TV (mm3) | 0.832 ± 0.045 | 0.820 ± 0.026 | 0.824 |
| BV/TV (ratio) | 0.276 ± 0.021 | 0.263 ± 0.016 | 0.641 | |
| Ct.Th (mm) | 0.141 ± 0.007 | 0.133 ± 0.008 | 0.514 | |
| Mid-diaphysis Tibiae | TV (mm3) | 0.477 ± 0.031 | 0.449 ± 0.029 | 0.539 |
| BV/TV (ratio) | 0.256 ± 0.018 | 0.239 ± 0.010 | 0.404 | |
| Ct.Th (mm) | 0.192 ± 0.009 | 0.186 ± 0.009 | 0.635 |
| Parameter | Wild Type | Nmur1 Knockout | p Value |
|---|---|---|---|
| Osteoid Surface (OS/BS, ratio) | 4.059 ± 1.361 | 8.400 ± 2.217 | 0.163 |
| Osteoclast Surface (OcS/BS, ratio) | 0.815 ± 0.309 | 0.608 ± 0.181 | 0.552 |
| Osteoclast Number (OcN/BS, #/mm2) | 0.036 ± 0.010 | 0.032 ± 0.007 | 0.764 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, Y.-T.; Manikowski, K.J.; Snyder, S.; Griffin, N.; Orr, A.L.; Hulsey, E.Q.; Born-Evers, G.; Zukosky, T.; Squire, M.E.; Hum, J.M.; et al. NMUR1 in the NMU-Mediated Regulation of Bone Remodeling. Life 2021, 11, 1028. https://doi.org/10.3390/life11101028
Hsiao Y-T, Manikowski KJ, Snyder S, Griffin N, Orr AL, Hulsey EQ, Born-Evers G, Zukosky T, Squire ME, Hum JM, et al. NMUR1 in the NMU-Mediated Regulation of Bone Remodeling. Life. 2021; 11(10):1028. https://doi.org/10.3390/life11101028
Chicago/Turabian StyleHsiao, Yu-Tin, Kelli J. Manikowski, Samantha Snyder, Nicole Griffin, Ashley L. Orr, Elizabeth Q. Hulsey, Gabriella Born-Evers, Tara Zukosky, Maria E. Squire, Julia M. Hum, and et al. 2021. "NMUR1 in the NMU-Mediated Regulation of Bone Remodeling" Life 11, no. 10: 1028. https://doi.org/10.3390/life11101028
APA StyleHsiao, Y.-T., Manikowski, K. J., Snyder, S., Griffin, N., Orr, A. L., Hulsey, E. Q., Born-Evers, G., Zukosky, T., Squire, M. E., Hum, J. M., Metzger, C. E., Allen, M. R., & Lowery, J. W. (2021). NMUR1 in the NMU-Mediated Regulation of Bone Remodeling. Life, 11(10), 1028. https://doi.org/10.3390/life11101028