The experimental system has been described previously [
10] and is shown schematically in
Figure 1. For these experiments, we always used the same gas mixture: 75 torr CO, 75 torr N
2, and 550 torr H
2 measured at room temperature, for a total initial gas pressure of 700 torr. The gas pressure increases as we heat the catalyst to our working temperature, then changes as the CO and H
2 are converted to various products during the course of the reaction. We note that we do not heat the tubing used to circulate the gas in this system, so that in many of the experiments, the pressure of water generated via the reactions in the system exceeds the equilibrium vapor pressure of water at room temperature, and measurements of the concentration of water in the system, therefore, reach a plateau at just under 20 torr. Condensation of the excess water vapor throughout the experimental system depletes the initial oxygen in the system and, therefore, could suppress the formation of CO
2. The FTIR spectrometer records the spectrum of the infrared active gases in the system at regular intervals, depending on the rate of change we expect for a particular experiment, and is set to provide a reasonably continuous record of the CO depletion and CH
4 generation as a function of reaction time for a given experiment.
We are aware that the pressures used in our experiments are much higher than those in the solar nebula and are also aware that this difference could affect the results of our experiments. We would love to duplicate nebular conditions and run our experiments continuously at nebular pressures for several hundred years, but such timescales are impractical. On average, our experiments run for from several days to several weeks. If an average experiment lasts for a week (6.05 × 105 s) but should last for a century (3 × 109 s), to duplicate conditions found in protostellar nebulae, we can obtain the same number of reactive CO collisions with our surfaces by increasing the pressure by a factor of ~5 × 103, from about 10−4 to 10−3 atmosphere in the nebula to between 0.5 and 5 atmosphere in our experiments. The experiments that we run at about one atmosphere, therefore, underestimate the number of collisions experienced by an average grain surface in the nebula in 100 years. Grains could circulate in such nebulae for 104 years or even longer (but at differing temperatures). We consider pressure to be a proxy for time and will generally ignore its effects on our products in these experiments. We have used this apparatus to carry out a variety of experiments to measure the characteristics of SMRs.
3.2. Carbon Gas to Solid Branching Ratios
Quantitative measurements of all gases in the system are made by generating synthetic spectra using the High-Resolution Transmission (HITRAN) spectroscopic database [
17] and comparing these data with experimentally measured FTIR spectra [
18]. These gas species include CO, CO
2, CH
4, C
2H
6, C
2H
2, NH
3, and H
2O. CO is the only source of carbon in the reaction system. Because we do not have a method to directly measure the number of moles of carbon deposited onto the catalyst surface, we measure this quantity indirectly by subtracting the molar quantities of CO, CO
2, CH
4, C
2H
2, and C
2H
6 in each spectrum from the initial number of moles of CO in the system and we assume that the difference was deposited onto the surface of the catalyst. Since CO
2, CH
4, C
2H
6, and C
2H
2 constitute the overwhelming majority of the carbonaceous gas phase products observed in our system, and the quantitative calculation of the gas phase concentrations from FTIR spectra are accurate to at least 10% or better, we believe that this assumption is reasonable. The fraction of the initial carbon available in the original carbon monoxide feedstock deposited on the grain surface can be calculated from this information.
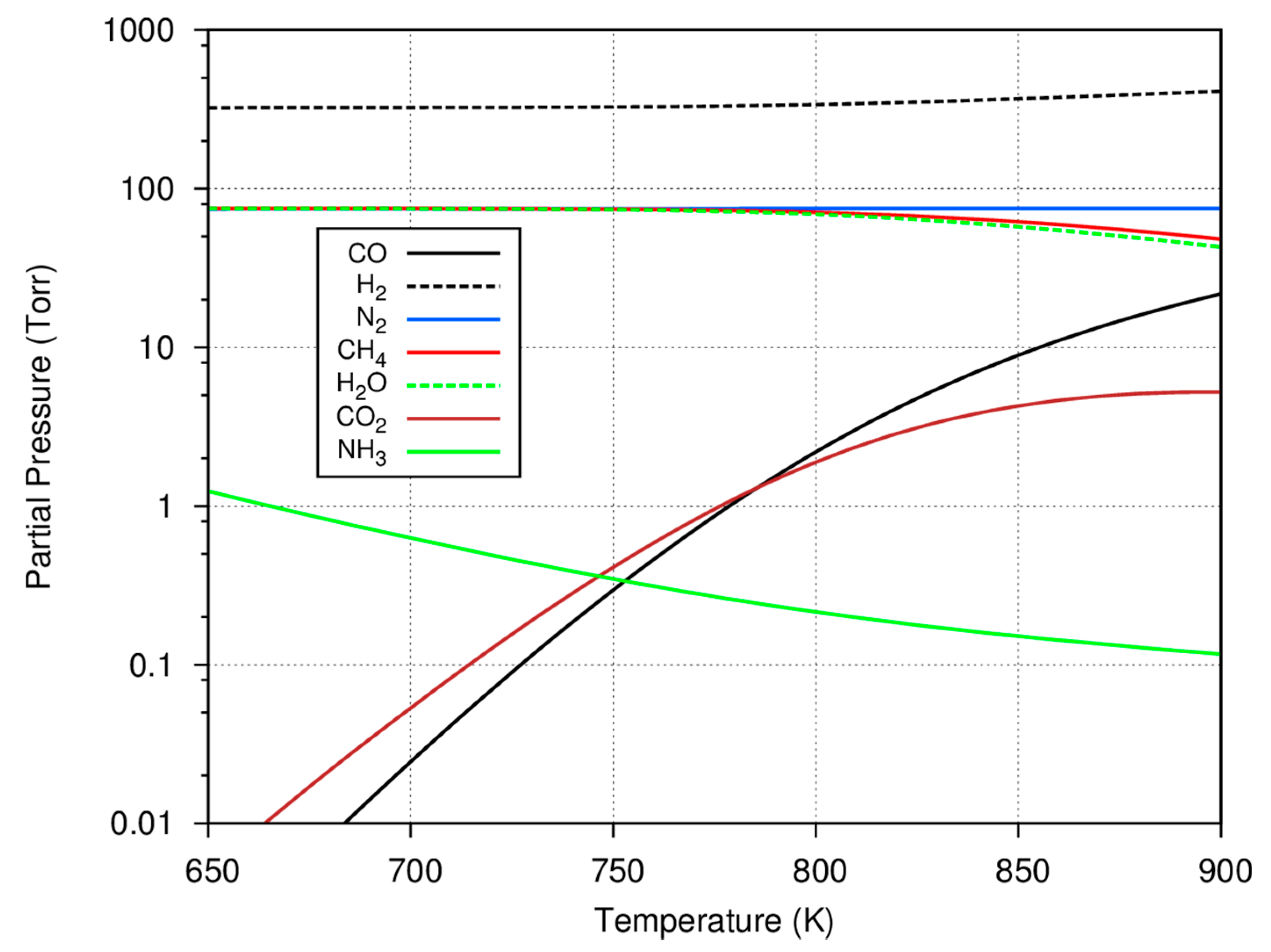
Before considering the experimental data, some insight can be gained by further considering what species are predicted to be present in an equilibrium mixture based on a typical initial gas charge. This information may be useful in assessing catalyst selectivity to the methanation reaction or the formation of deposited carbon and hydrocarbons. To compute the equilibrium gaseous concentrations with such multiple reactions, the minimization of Gibbs free energy technique using Lagrange multipliers was used [
19] with a starting gas mixture of 75 Torr carbon monoxide, 75 Torr nitrogen, and 550 Torr hydrogen. The results of these calculations for the major reactant and product gases are shown as a function of temperature in
Figure 2.
As shown in
Figure 2, the formation of methane is favored at lower temperatures, with virtually all of the carbon monoxide consumed and converted to methane. As the temperature increases, the reaction shifts, less methane is predicted to be formed, and there is residual carbon monoxide as well as some carbon dioxide in the equilibrium mixture. Since the consumption of carbon monoxide is used to determine the progress of the experiments, it is important to note that not all of the carbon monoxide may be consumed at the higher system temperatures. Because of the low system pressure (compared to those used in the industrial Haber–Bosch process), only a small amount of ammonia is predicted to be formed and larger, equilibrium amounts of ammonia are also favored at the lower temperatures. In several experiments, it is common to form ~50–60 Torr of methane; in these cases, it is clear that the overall reaction at equilibrium favors the methanation reaction and the selectivity to form condensed hydrocarbons is low.
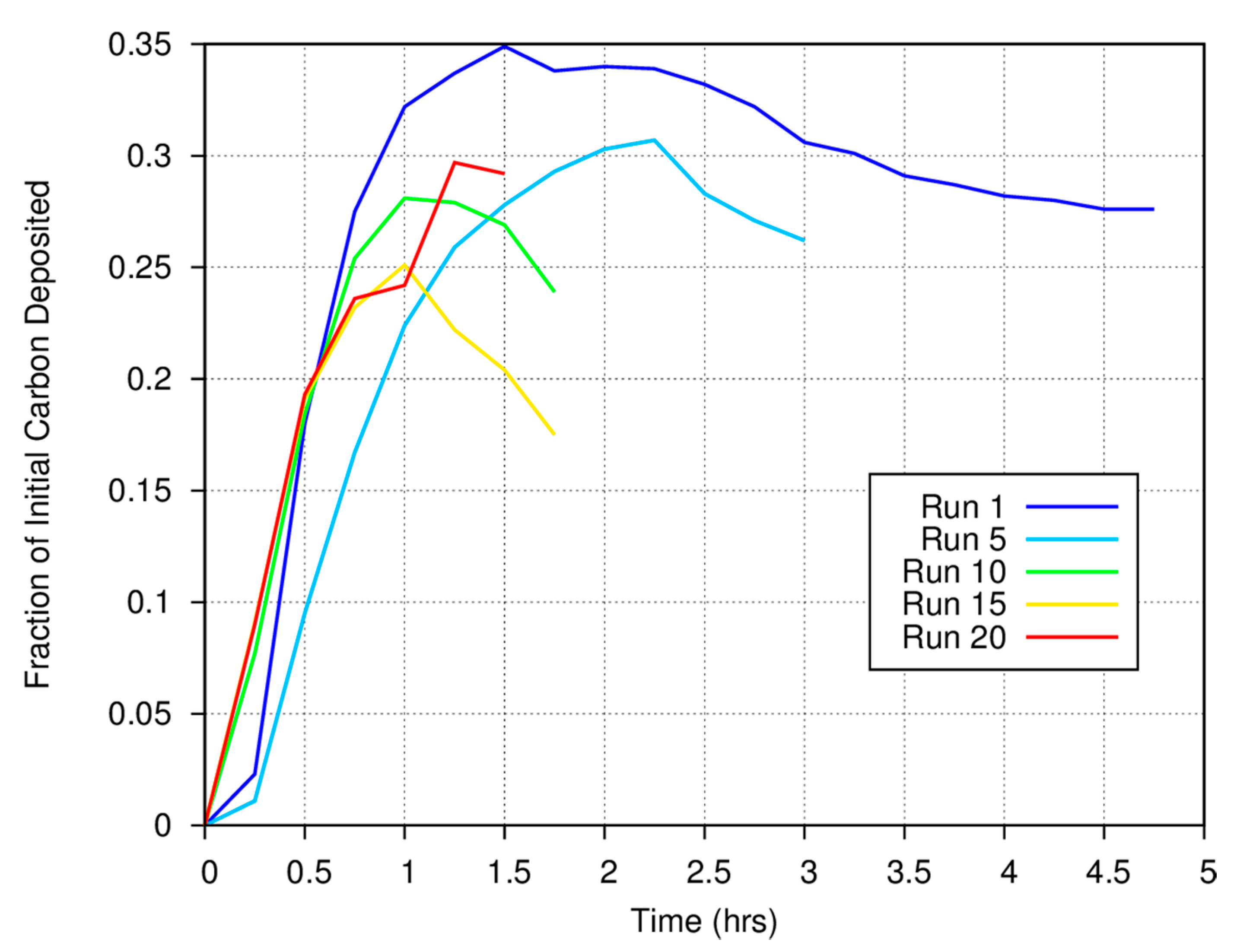
Figure 3 shows the measured carbon gas/solid fraction for a magnetite catalyst as a function of time and run number for a series of experiments conducted at 773 K. Approximately 25% of the carbon in the initial CO is deposited onto the surface of the Fe
3O
4 catalyst by the end of each experimental run, while the bulk of the carbon remains as gas phase CO, CO
2, CH
4, and C
2H
6. Note the anomalous behavior at the start of runs 1 and 5. We always measure the initial CO concentration and perform quantitative measurements using the synthetic spectra generated using the HITRAN database at the temperature of the FTIR cell (room temperature) and the measured total system pressure. The anomaly results because there is a rapid rise in total system pressure when heat is applied to the catalytic “finger”, thus increasing the concentration of gas phase species in the FTIR cell.
For the first measurement made at the start of a run, the pressure will be 700 torr (by design at room temperature). However, because the catalyst finger is now being heated to 773 K (for the data shown in
Figure 3) as we start the system, the temperature of the finger will be somewhat higher, thus increasing the pressure throughout the closed system. A higher pressure in the system will lead to an artificial increase in the gas phase abundance of carbon as determined from comparison between synthetic and measured spectra for the second and all subsequent measurements until the surface-mediated reactions begin to deposit carbon onto the grains or until fewer moles of gas phase species are formed as reaction products. If carbon deposition or the overall reaction rates are slow, the negative anomaly will persist for some time. For faster rates of deposition, carbon will be taken out of the gas phase more rapidly and increasing gas pressure can be compensated for by carbon deposition. Note that in
Figure 3, the anomaly is pronounced for run 1, less pronounced in run 5, and much less obvious in runs 10, 15, and 20. As the catalytic finger was heated at the same rate for each experimental run, these data imply either that the rate at which carbon is deposited onto the surface of the catalyst at the beginning of each experiment increased as a function of run number or that the overall reaction rate increased. Thus, the more carbon that has already been deposited onto the magnetite surface, the faster carbon is deposited onto that surface in subsequent experiments.
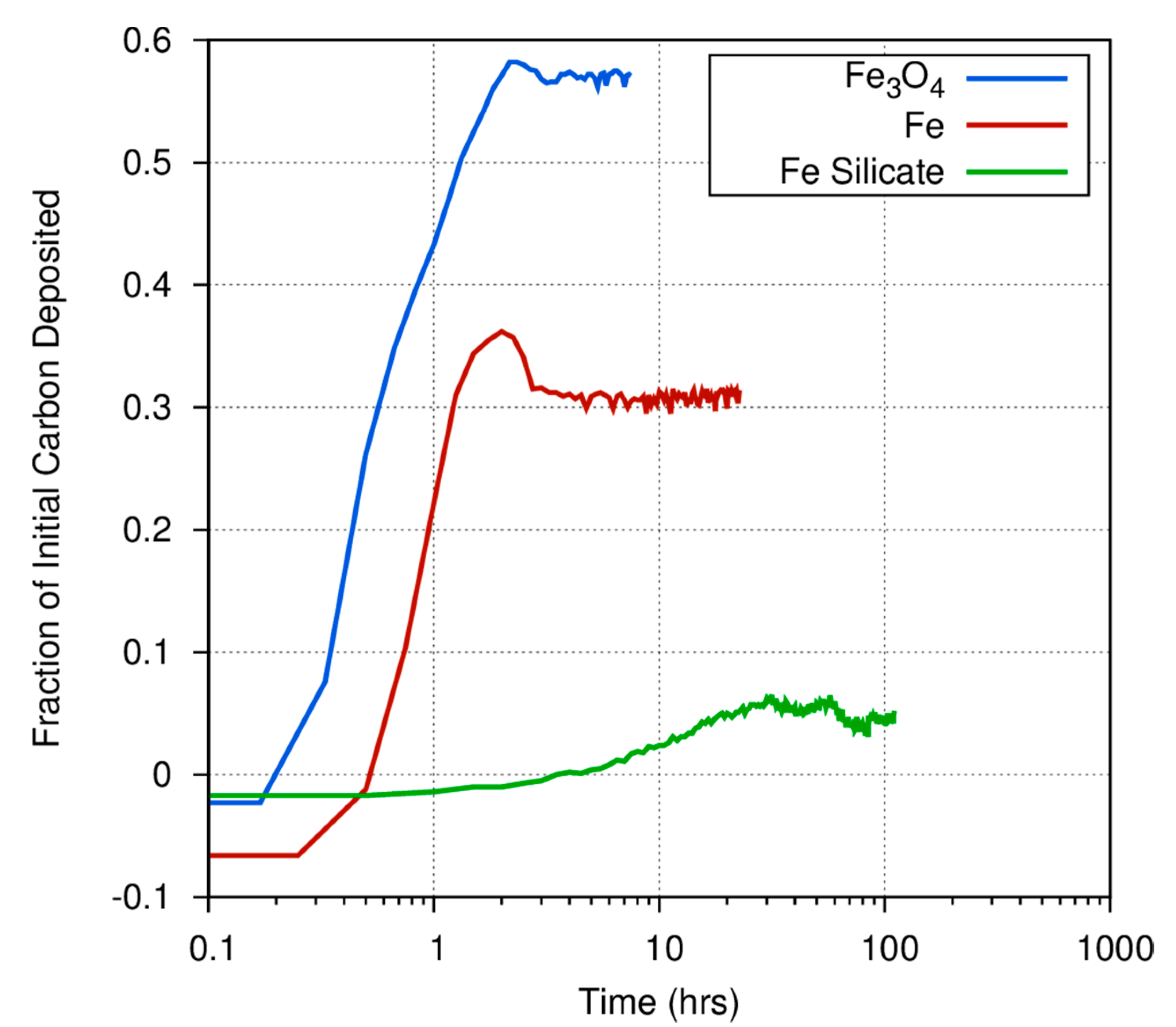
Figure 4 shows the different initial rate and gas/solid fraction for carbon deposition onto iron, magnetite, and amorphous iron silicate smokes at 873 K. Fe
3O
4 very efficiently converts the initial C in the CO feed stock into a carbonaceous deposit, as compared to pure Fe or the amorphous silicate smokes.
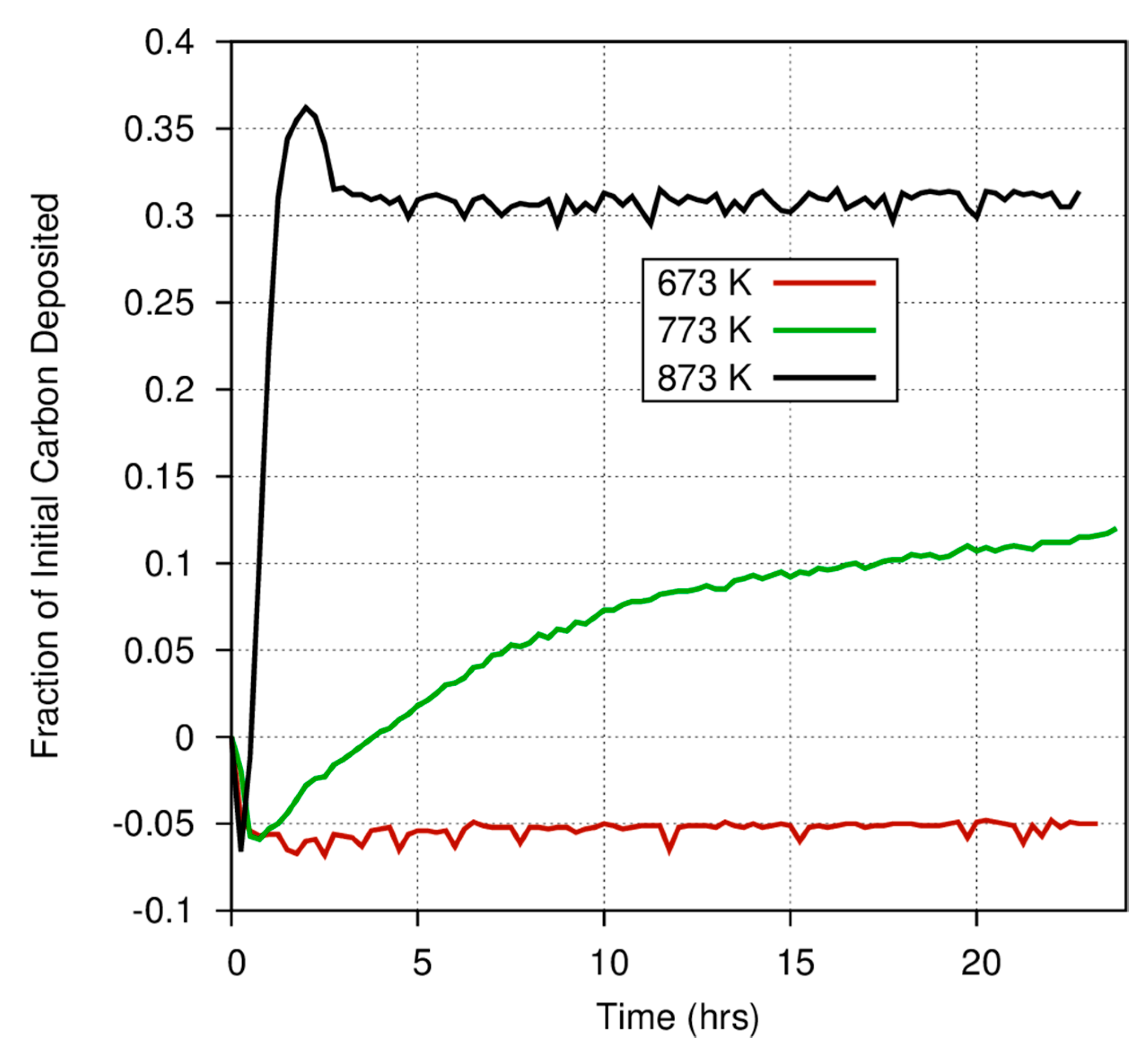
Figure 4 also demonstrates the considerable differences among catalysts in the fraction of carbon deposited onto grain surfaces versus the fraction that is converted into gas phase products. At 873 K, nearly 60% of the CO exposed to a magnetite catalyst is deposited onto the surface, whereas only 30% of the carbon exposed to an iron catalyst is converted into a carbonaceous deposit. Even at high temperatures, where the Boudouard reaction should become much more important, much less CO is converted into a solid on the amorphous iron silicate catalysts. The effect of temperature is more dramatically illustrated in
Figure 5, where the fraction of CO deposited onto an iron catalyst surface at three different temperatures varies from 0.3 at 873 K to approximately 0.12 (accounting for the anomaly discussed above) at 773 K, and down to ~0.05 at 673 K. Obviously, for a pure Fe catalyst, the branching ratio of the carbon deposited onto the grain surface compared to the carbon incorporated into volatile compounds is a very strong function of temperature. Higher temperature yields fewer volatiles. This is consistent with the general trend in the Boudouard reaction but is not consistent with thermodynamic predictions of the instability of free carbon in the presence of excess hydrogen. More information on these experiments is available in Nuth et al. [
20].
3.3. Surface Area and Morphology of the Carbonaceous Solids from SMRs
Surface-mediated reactions can occur at active catalytic sites on almost any grain surface [
9,
10] and we have previously demonstrated that the deposition of refractory carbonaceous material on grain surfaces can actually enhance the rate of such reactions [
11]. We had made the simple assumption that the carbonaceous coating grew to cover the surface of the underlying catalyst, leaving an outer coating that completely and uniformly covered the original grain. Such “grain varnishes” or refractory organic residues have frequently been described in the past as the result of ultraviolet or radiation processing of ices containing simple organic precursors such as CH
4, CH
3OH, CH
2O, and NH
3 [
21,
22]. It seemed natural to assume that a similar morphology would result from surface-mediated processes. That assumption is incorrect.
We used a Quantichrome Nova E series surface area analyzer and the Brunauer–Emmett–Teller (BET) technique [
23] with N
2 as the adsorbed gas to measure the area of our initial pure magnetite catalyst, as well as small samples of the catalyst that had been used for 5, 10, 15, and 20 experimental runs. We found that the area of our catalyst increased significantly at each measurement. This implies that the area increases during each individual run as well, making quantification of the reaction kinetics as a function of the catalytic surface area an interesting challenge that we will tackle in future work. The results of these measurements for an initially pure magnetite catalyst are given in
Table 1.
As can be seen, the active catalytic surface area more than doubled relative to the surface area of the original magnetite catalyst within the first five experimental runs, then increased to more than four times the initial magnetite surface area after 10 runs and to more than six times the original area after 20 experiments. It is highly unlikely that the catalyst remains pure magnetite as its surface area increases.
Unfortunately, these results should be taken as lower limits to the actual increase in surface area that occurred during these experiments. As we began to quantitatively measure the reaction rates per unit surface area of catalyst, we became aware that the rates did not scale by the area of the catalyst as we had expected. Instead, the rate for iron powders compared to the rate for several lengths of iron wires indicated that most of the iron powder was not seen by the reactive gas mixture. The experimental system shown in
Figure 1 was set up as a fluidized bed reactor, in which the gas injected at the bottom of the catalyst bed “liquefies” the very fluffy iron silicate smokes that we first tested as catalysts. In such a system, the gas interacts freely with the entire catalyst bed. Use of the much denser iron, hematite, or magnetite powders in this system eliminates the fluidity of the catalyst at the gas flow rates available in our system and even though we thought that the gas permeated the entire reaction bed, it became obvious to us that a significant portion of the iron powder catalyst did not participate in the reaction. While this does not change the product distribution of the reactants, it certainly effects measurements of the reaction kinetics and means that measurements of the surface area per gram of an iron or magnetite powder catalyst, measured for a randomly drawn sample of the bulk material, averaged the increased surface area of active grains with a larger quantity of grains that had minimal interaction with the gas.
Even this lower limit for the rapid increase in the surface area of the catalyst was very difficult to explain based on our previous experiments and with our ingrained assumptions concerning the morphology of the deposited coating. Previous analyses of the coating on iron silicate smokes with initial surface area ~125 m
2/g and a total mass less than 5 g showed that after 20 runs, the grains were 10% by mass carbon and 0.2% by mass nitrogen [
24]. Increasing the surface area of our catalyst by a factor of six would require more than doubling the average radius of the 325 mesh magnetite catalyst and would require much more than the available quantity of carbon in the system to accomplish this increase. Based on our previous work, we would expect that much less than a gram of carbon deposition occurs (a total on all grains in the system) over 20 runs and we know that a significant fraction of the available carbon is converted into CO
2, CH
4, and other volatile organic species via the overall reaction [
11]. This does not leave enough mass to coat the initial magnetite grains to sufficient thickness to increase their surface area by factors of four or more. Continuous carbonaceous coatings on the catalytic surfaces can therefore be ruled out by these measurements—a more complex surface morphology is required.
In previous experiments conducted at 873 K using a graphite catalyst, we have shown the formation and growth of carbon nanotubes [
12], essentially a thin string of carbon with a very high surface area per unit mass. A TEM image of these deposits is shown in
Figure 6 to illustrate the very large surface area possible for such morphologies that we had believed were unique to experiments using graphitic catalysts and their large reservoir of potentially reactive carbon. We had never seen evidence for nanotube formation at temperatures less than 823 K for any other catalyst, including in both SEM analyses as well as in electron diffraction analyses of lower temperature grain coatings. In our previous SEM images, the surface always appeared to be “lumpy”—which we attributed to grain clumping and other macroscopic features produced during the reaction.
Higher resolution SEM images of iron silicate smokes used as catalysts at 873 K tell a very different story, as can be seen in
Figure 7. It appears that active regions on the surface of the catalyst initially promote carbon deposition in the local area. However, rather than spreading uniformly over the surface of the grains, the carbon deposit also begins to grow away from the catalyst surface. While, in some ways, this initially appears to resemble VLS (Vapor–Liquid–Solid) growth along a crystalline “c-axis”, the resultant deposit is neither crystalline nor is it a smooth needle or whisker—in fact, it is rather clumpy and quite irregular. It also appears that much, if not all, of the deposited carbon is capable of promoting additional surface-mediated reactions and further carbon deposition much as we had observed when using the graphite catalyst. The newly deposited carbon does not appear to thicken the nanotubes themselves, but instead, appears to cause them to lengthen. Based on experiments on the formation and growth of nanotubes [
12], we know that carbon atoms are relatively easily mobilized from such initial condensates to form longer nanotubes—another similarity to VLS growth. It is possible that once such tubes begin to form, the chemical composition of the original catalyst becomes increasingly less important with time.
The formation of filamentous carbon deposits is one possibility that may explain both the dramatic increase in specific surface area and the increased catalytic activity. Such filamentous carbon is known to occur in carburizing and reducing atmospheres in the 400–800 °C range. Such deposits have been known for some time; one of the first reports of such deposits was made after observations of the reaction of carbon monoxide with iron oxide on the brickwork in a blast furnace [
26]. Schulz et al. [
27] found such structures while studying Fischer–Tropsch and methanation catalysts and termed the deposits “carbon expanded iron”. It was suggested that, under certain conditions, the carbon deposition broke the bulk metal into finer particles [
27]. The carbon would grow into filaments that would keep the metal particles from agglomerating, thereby making the metal more accessible to reactant gases. Schulz et al. [
26] noted the unexpected catalytic activity per gram of material after the growth of these carbon-expanded iron filaments. Such behavior may be responsible for the increased catalytic activity noted with the current catalysts, where reaction rates increase with successive runs. However, in our initial experiments (e.g.,
Figure 7), the underlying catalytic dust is amorphous iron silicate rather than iron metal.
In industrial processes, such carbonaceous deposits can occur on the steel reactor walls, causing harmful effects. One particular growth of carbon, called “metal dusting”, causes the corrosion of vessel walls, resulting in a dust of graphite and small metal particles [
28]. Both the filamentous carbon and metal dusting growth occur with metals that absorb carbon, e.g., iron, nickel, and cobalt. Carbon is absorbed and transported within the metal due to a gradient in carbon activity. In fact, the diffusion of carbon in the catalyst particle is thought to be the rate-determining step in the reaction sequence because there is very close agreement between activation energies for filament growth and those for diffusion of carbon through the metals [
29].
The formation of filamentous carbon deposits on metal particles is outlined in six steps given by Bonnet et al. [
30]. These steps include: (1) the decomposition of hydrocarbons on the metal surface and supersaturation of the metal with absorbed carbon; (2) the nucleation and growth of cementite, Fe
3C; (3) carbon diffusion becomes impeded through the cementite layer and hinders further transfer from the gas phase; (4) graphite begins to precipitate on the cementite surface, reducing the activity of carbon at the graphite/cementite interface and thus, making cementite unstable and causing it to decompose into carbon and iron; (5) these free carbon atoms attach to the basal planes of graphite that grow into the cementite while the iron atoms agglomerate to form small particles; and finally, (6) these small particles are able to act as catalysts for further carbon deposition and growth. While these steps outline the growth of filamentous carbon with metallic iron, Bonnet et al. proposed a new mechanism whereby iron oxides can also form such filamentous structures by being converted directly to cementite without having metallic iron as an intermediate phase [
30]. This filamentous carbon should thus be able to form with iron oxide mixtures as well as on metallic iron covered with oxide layers. Thus far, the experimental evidence seems to support the formation of this filamentous carbon and further experiments will be focused on imaging and chemical analyses of such deposits using high resolution microscopy to investigate this possibility.
3.4. Implications of Filamentous Carbon Solids for Nebular Processes
Our experiments indicate that macroscopic carbon particles, many hundreds—or even thousands—of nanometers in length and quite irregular in shape, could form at least on the surfaces of amorphous iron and magnesium silicates, iron, magnetite and graphite grains, and probably on many other surfaces as well. This could have several consequences. First, such carbon-rich growth could at least partially replenish some of the presolar, graphite-like dust previously destroyed in high temperature nebular shocks, lightning plasma tubes, or in grain aggregates heated while in close proximity to the early sun. While we know that a large fraction of presolar silicate grains were processed in the nebula to produce the much larger dust particles found in meteorite matrix, presolar carbon dust must have experienced similar heating processes, though no quantitative measure of such processing is yet available in the literature. However, while silicates are converted into liquid droplets or vapor at high temperatures—and silicate vapor can recondense—carbon is converted into CO or CO2 at high temperatures in the oxygen-rich solar nebula or when in direct contact with silicate grains. Surface-mediated reactions would be a significant pathway to convert such gases back into the solid carbon species found in meteorites.
An interesting possibility, if such processes occur on the surfaces of larger (mm-scale) silicate and metallic grains in the nebula, is that such growth could act much in the same manner as do fresh snowy condensates on the surfaces of the larger ice grains in Saturn’s rings. An example of this is shown in
Figure 8, where a section of iron wire is compared before and after use as a catalyst for a single experimental run [
20]. Such fluffy grain coatings could damp out the momentum in gently colliding particles, changing the coefficient of restitution and promoting grain–grain sticking [
31,
32]. Surface-mediated reactions would happen fastest in the higher temperature, higher pressure regions of the inner solar nebula, just where chondrules, CAIs, and other macroscopic meteoritic components are also forming. Under such circumstances, the solid carbonaceous products would tend to be more graphitic and could even be similar to the stacked-cup nanotubes that we have observed previously [
12]. If some of these refractory surfaces served as catalytic sites for the growth of filamentous “whiskers”, then such surface growth could also promote the aggregation of these larger meteoritic components. In fact, at least one study has reported an association between carbon nanotubes and CAIs [
33]. Such carbonaceous growth could have been the “Velcro” that promoted the aggregation of meteorite parent bodies in the high temperature inner nebula.
Finally, we have previously suggested that “Fischer–Tropsch-Type” surface-mediated reactions could convert CO or CO
2 produced in high temperature nebular events back into carbonaceous gases and solids [
34]. These processes would have two major consequences. First, the conversion of CO into more reactive carbonaceous species breaks down the assumed stable C
16O reservoir that is required for the CO self-shielding mechanism [
35,
36,
37,
38] to enrich the solar nebula in heavier oxygen isotopes and produce mass independent oxygen isotopic fractionation in such meteoritic components as CAIs and chondrules [
39,
40]. Second, solid carbon fibers are much more easily incorporated into growing planetesimals in the warm inner solar nebula than would be the host of gaseous hydrocarbons generated by SMRs [
10]. These carbonaceous solids could serve as a reactive feedstock to synthesize a wide range of organic biomolecules as they react in the presence of water and amorphous magnesium and iron silicates in evolving planetesimals. We are not claiming here that SMR reaction products constitute the single—or even the most important—mechanism that forms the full distribution of organic molecules in asteroids or comets or that are found in meteorites or interplanetary dust particles. There are many different processes that can be important sources for particular compounds in many different meteorite types [
41]. However, we do believe that surface-mediated reactions are a very efficient mechanism for converting nebular CO or CO
2 into solid carbonaceous materials and the products of such reactions have not yet been investigated.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}