The Ancient History of Peptidyl Transferase Center Formation as Told by Conservation and Information Analyses

 ,
,  ,
, 
Abstract
1. Introduction
2. Material and Methods
2.1. Download of Complete 23S Ribosomal RNAs from Public Databases
2.2. Retrieving PTCs from 23S Ribosomal RNA Sequences
2.3. Multiple Alignment, Filtering, and Production of PTC Datasets
2.4. Information Theory Analysis of PTCs
2.5. Mapping Conservation into 2D and 3D PTC Structures
2.6. 3D Modeling of the Different PTC Sequences and Structural Comparisons
3. Results
3.1. PTC Datasets: Production and Taxonomic Analysis
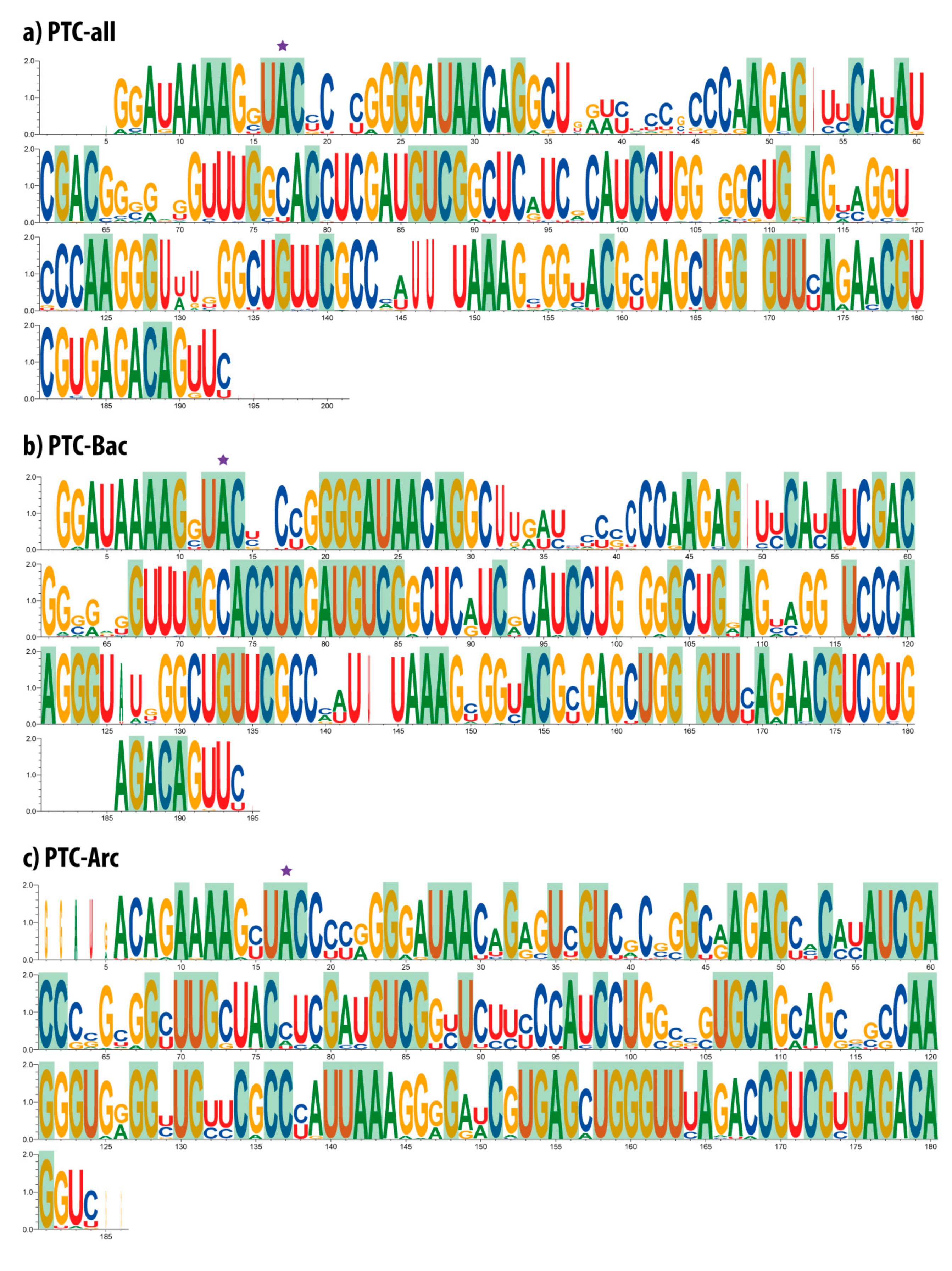
3.2. Multiple Alignment and Sequence Conservation of PTC Datasets
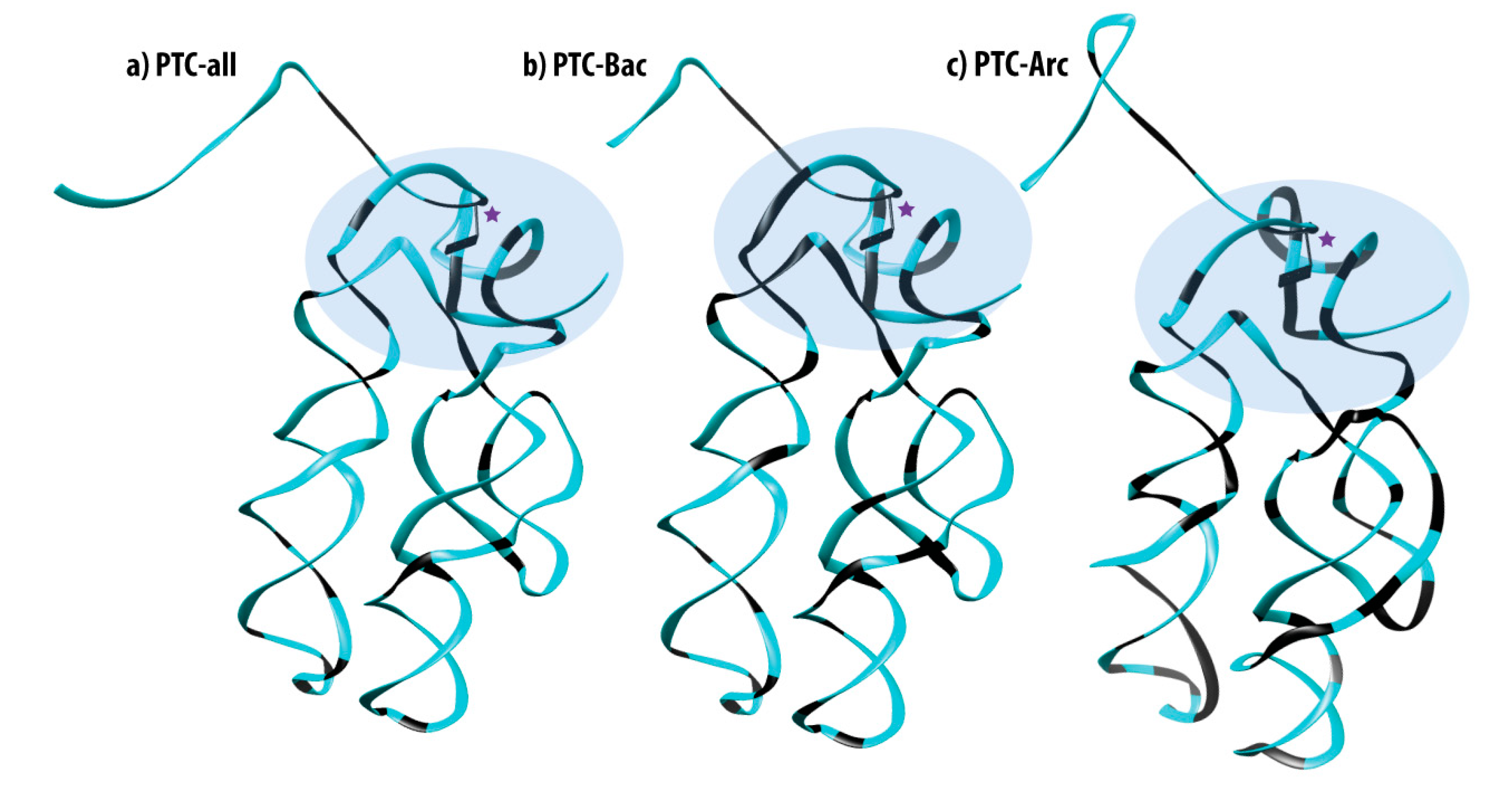
3.3. Mapping 100% Conserved Sites into the 3D Structure of the PTC
3.4. Identity Elements, Entropy Variation, Information Variation
3.5. Mapping Identity Elements and Information Clusters into the 2D Structure of the PTC
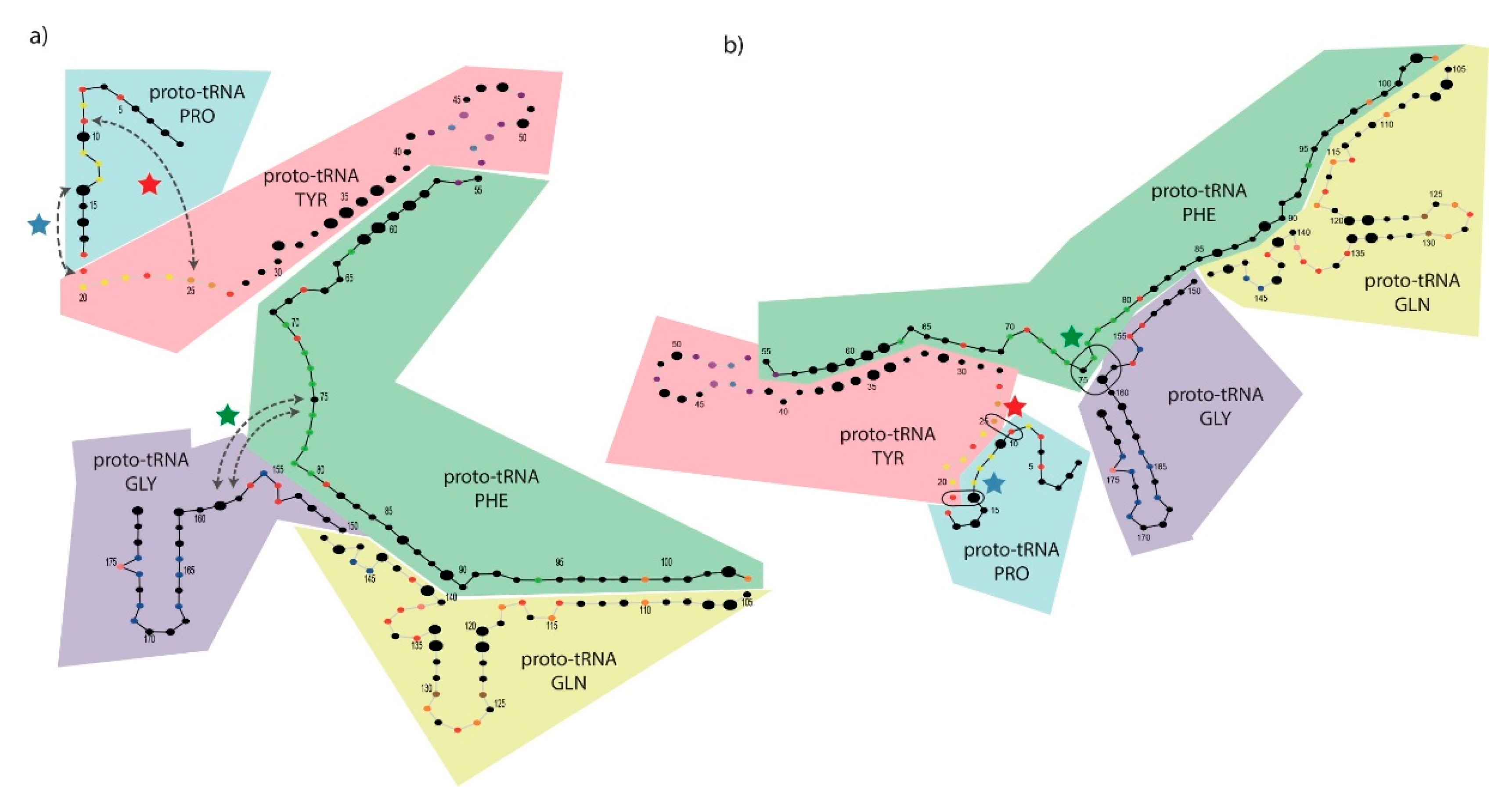
3.6. Mapping Proto-tRNAs into the 2D Structure of the PTC
3.7. Mapping Identity Elements and Information Clusters into the 3D Structure of the PTC
3.8. Structural Alignment of PTC Datasets to T. Thermophilus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Noller, H.F.; Woese, C.R. Secondary structure of 16S ribosomal RNA. Science 1981, 212, 403–411. [Google Scholar] [CrossRef]
- Noller, H.F.; Hoffarth, V.; Zimniak, L. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 1992, 256, 1416–1419. [Google Scholar] [CrossRef]
- Lohse, P.A.; Szostak, J.W. Ribozyme-catalysed amino-acid transfer reactions. Nature 1996, 381, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Lanier, K.A.; Petrov, A.S.; Williams, L.D. The Central Symbiosis of Molecular Biology: Molecules in Mutualism. J. Mol. Evol. 2017, 85, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Vitas, M.; Dobovišek, A. In the Beginning was a Mutualism—On the Origin of Translation. Orig. Life Evol. Biosph. 2018, 48, 223–243. [Google Scholar] [CrossRef] [PubMed]
- De Farias, S.T.; Prosdocimi, F. A Emergência dos Sistemas Biológicos: Uma Visão Molecular da Origem da Vida, 1st ed.; ArtecomCiencia: Rio de Janeiro, Brazil, 2019; ISBN 978-65-900624-1-3. [Google Scholar]
- Prosdocimi, F.; José, M.V.; de Farias, S.T. The First Universal Common Ancestor (FUCA) as the Earliest Ancestor of LUCA’s (Last UCA) Lineage. In Evolution, Origin of Life, Concepts and Methods; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 43–54. ISBN 978-3-030-30363-1. [Google Scholar]
- Bokov, K.; Steinberg, S.V. A hierarchical model for evolution of 23S ribosomal RNA. Nature 2009, 457, 977–980. [Google Scholar] [CrossRef]
- Petrov, A.S.; Bernier, C.R.; Hsiao, C.; Norris, A.M.; Kovacs, N.A.; Waterbury, C.C.; Stepanov, V.G.; Harvey, S.C.; Fox, G.E.; Wartell, R.M.; et al. Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 10251–10256. [Google Scholar] [CrossRef]
- Caetano-Anollés, G. Ancestral Insertions and Expansions of rRNA do not Support an Origin of the Ribosome in Its Peptidyl Transferase Center. J. Mol. Evol. 2015, 80, 162–165. [Google Scholar] [CrossRef]
- Petrov, A.S.; Williams, L.D. The ancient heart of the ribosomal large subunit: A response to Caetano-Anolles. J. Mol. Evol. 2015, 80, 166–170. [Google Scholar] [CrossRef]
- Rodnina, M.V. The ribosome as a versatile catalyst: Reactions at the peptidyl transferase center. Curr. Opin. Struct. Biol. 2013, 23, 595–602. [Google Scholar] [CrossRef]
- Caetano-Anollés, G.; Caetano-Anollés, D. Computing the origin and evolution of the ribosome from its structure—Uncovering processes of macromolecular accretion benefiting synthetic biology. Comput. Struct. Biotechnol. J. 2015, 13, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J. Induced fit of the peptidyl-transferase center of the ribosome and conformational freedom of the esterified amino acids. RNA 2016, 23, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Agmon, I.; Bashan, A.; Zarivach, R.; Yonath, A. Symmetry at the active site of the ribosome: Structural and functional implications. Biol. Chem. 2005, 386, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Belousoff, M.J.; Davidovich, C.; Zimmerman, E.; Caspi, Y.; Wekselman, I.; Rozenszajn, L.; Shapira, T.; Sade-Falk, O.; Taha, L.; Bashan, A.; et al. Ancient machinery embedded in the contemporary ribosome. Biochem. Soc. Trans. 2010, 38, 422–427. [Google Scholar] [CrossRef]
- Tamura, K. Ribosome evolution: Emergence of peptide synthesis machinery. J. Biosci. 2011, 36, 921–928. [Google Scholar] [CrossRef]
- Caetano-Anollés, G.; Sun, F.-J. The natural history of transfer RNA and its interactions with the ribosome. Front. Genet. 2014, 5, 5. [Google Scholar]
- De Farias, S.T.; Rêgo, T.G.; José, M.V. Origin and evolution of the Peptidyl Transferase Center from proto-tRNAs. FEBS Open Bio 2014, 4, 175–178. [Google Scholar] [CrossRef]
- Root-Bernstein, M.; Root-Bernstein, R. The ribosome as a missing link in the evolution of life. J. Theor. Biol. 2015, 367, 130–158. [Google Scholar] [CrossRef]
- De Farias, S.T.; Rêgo, T.G.; José, M.V. Peptidyl Transferase Center and the Emergence of the Translation System. Life 2017, 7, 21. [Google Scholar] [CrossRef]
- Demongeot, J.; Seligmann, H. Evolution of tRNA into rRNA secondary structures. Gene Rep. 2019, 17, 100483. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 48, D84–D86. [Google Scholar]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, 00, 2.3.1–2.3.22. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Stairs, C.W.; Dombrowski, N.; Eme, L.; Lombard, J.; Caceres, E.F.; Greening, C.; Baker, B.J.; Ettema, T.J.G. Proposal of the reverse flow model for the origin of the eukaryotic cell based on comparative analyses of Asgard archaeal metabolism. Nat. Microbiol. 2019, 4, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; Van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Zamudio, G.S.; José, M.V. Identity Elements of tRNA as Derived from Information Analysis. Orig. Life Evol. Biosph. 2017, 48, 73–81. [Google Scholar] [CrossRef]
- Meilă, M. Comparing Clusterings by the Variation of Information. In Learning Theory and Kernel Machines: 16th Annual Conference on Learning Theory and 7th Kernel Workshop, COLT/Kernel 2003, Washington, DC, USA, 24–27 August 2003: Proceedings; Springer: Berlin/Heidelberg, Germany, 2003; pp. 173–187. ISBN 978-3-540-40720-1. [Google Scholar]
- Zamudio, G.S.; Palacios-Pérez, M.; José, M.V. Information theory unveils the evolution of tRNA identity elements in the three domains of life. Theory Biosci. 2020, 139, 77–85. [Google Scholar] [CrossRef]
- Bernier, C.R.; Petrov, A.S.; Waterbury, C.C.; Jett, J.; Li, F.; Freil, L.E.; Xiong, X.; Wang, L.; Migliozzi, B.; Hershkovits, E.; et al. RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss. 2014, 169, 195–207. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Rother, K.; Puton, T.; Bujnicki, J.M. ModeRNA: A tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 2011, 39, 4007–4022. [Google Scholar] [CrossRef]
- Gong, S.; Zhang, C.; Zhang, Y. RNA-align: Quick and accurate alignment of RNA 3D structures based on size-independent TM-scoreRNA. Bioinformatics 2019, 35, 4459–4461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Prosdocimi, F.; Linard, B.; Pontarotti, P.; Poch, O.; Thompson, J.D. Controversies in modern evolutionary biology: The imperative for error detection and quality control. BMC Genom. 2012, 13, 5. [Google Scholar] [CrossRef]
- Schroeder, R.; Barta, A.; Semrad, K. Strategies for RNA folding and assembly. Nat. Rev. Mol. Cell Biol. 2004, 5, 908–919. [Google Scholar] [CrossRef]
- Schultes, E.A.; Bartel, D.P. One sequence, two ribozymes: Implications for the emergence of new ribozyme folds. Science 2000, 289, 448–452. [Google Scholar] [CrossRef]
- Brion, P.; Westhof, E. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 113–137. [Google Scholar] [CrossRef]
- Draper, D.E. A guide to ions and RNA structure. RNA 2004, 10, 335–343. [Google Scholar] [CrossRef] [PubMed]
- De Farias, S.T. Suggested phylogeny of tRNAs based on the construction of ancestral sequences. J. Theor. Biol. 2013, 335, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Mörl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Pütz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef] [PubMed]
- Korostelev, A.; Trakhanov, S.; Laurberg, M.; Noller, H.F. Crystal Structure of a 70S Ribosome-tRNA Complex Reveals Functional Interactions and Rearrangements. Cell 2006, 126, 1065–1077. [Google Scholar] [CrossRef]
- Krupkin, M.; Matzov, D.; Tang, H.; Metz, M.; Kalaora, R.; Belousoff, M.J.; Zimmerman, E.; Bashan, A.; Yonath, A. A vestige of a prebiotic bonding machine is functioning within the contemporary ribosome. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2972–2978. [Google Scholar] [CrossRef]
- De Farias, S.T.; José, M.V. Transfer RNA: The molecular demiurge in the origin of biological systems. Prog. Biophys. Mol. Boil. 2020, 153, 28–34. [Google Scholar] [CrossRef]
- Prosdocimi, F.; de Farias, S.T. From FUCA To LUCA: A Theoretical Analysis on the Common Descent of Gene Families. Acta Sci. Microbiol. 2020, 3, 1–9. [Google Scholar]
- Ivanov, V.I.; Bondarenko, S.A.; Zdobnov, E.M.; Beniaminov, A.D.; Minyat, E.E.; Ulyanov, N.B. A pseudoknot-compatible universal site is located in the large ribosomal RNA in the peptidyltransferase center. FEBS Lett. 1999, 446, 60–64. [Google Scholar] [CrossRef]
- Polacek, N.; Mankin, A.S. The Ribosomal Peptidyl Transferase Center: Structure, Function, Evolution, Inhibition. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 285–311. [Google Scholar] [CrossRef]
- Chirkova, A.; Erlacher, M.D.; Clementi, N.; Żywicki, M.; Aigner, M.; Polacek, N. The role of the universally conserved A2450–C2063 base pair in the ribosomal peptidyl transferase center. Nucleic Acids Res. 2010, 38, 4844–4855. [Google Scholar] [CrossRef]
- Davidovich, C.; Belousoff, M.J.; Wekselman, I.; Shapira, T.; Krupkin, M.; Zimmerman, E.; Bashan, A.; Yonath, A. The Proto-Ribosome: An ancient nano-machine for peptide bond formation. Isr. J. Chem. 2010, 50, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Hayashi, G.; Katoh, T.; Suga, H. An orthogonal ribosome-tRNA pair via engineering of the peptidyl transferase center. Nat. Chem. Biol. 2014, 10, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J. Physico-chemical Constraints Connected with the Coding Properties of the Genetic System. J. Theor. Biol. 2000, 202, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Cibils, M.; Libchaber, A. Emergence of a Code in the Polymerization of Amino Acids along RNA Templates. PLoS ONE 2009, 4, e5773. [Google Scholar] [CrossRef] [PubMed]
- Zaucha, J.; Heddle, J.G. Resurrecting the Dead (Molecules). Comput. Struct. Biotechnol. J. 2017, 15, 351–358. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Name | Clades | Number of PTC Sequences | Alignment Size (Gaps) | Positions 100% Conserved |
|---|---|---|---|---|
| PTC-all | Bacteria Archaea Eukarya | 1424 | 201 (10) | 42 |
| PTC-Bac | Bacteria * | 1301 | 195 (8) | 62 |
| PTC-Arc | Archaea * | 118 | 186 (7) | 83 |
| PTC-Pro | Proteobateria * | 564 | 186 (7) | 110 |
| PTC-Fir | Firmicutes ** | 237 | 184 (4) | 122 |
| PTC-Act | Actinobacteria | 153 | 179 (0) | 132 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prosdocimi, F.; Zamudio, G.S.; Palacios-Pérez, M.; Torres de Farias, S.; V. José, M. The Ancient History of Peptidyl Transferase Center Formation as Told by Conservation and Information Analyses. Life 2020, 10, 134. https://doi.org/10.3390/life10080134
Prosdocimi F, Zamudio GS, Palacios-Pérez M, Torres de Farias S, V. José M. The Ancient History of Peptidyl Transferase Center Formation as Told by Conservation and Information Analyses. Life. 2020; 10(8):134. https://doi.org/10.3390/life10080134
Chicago/Turabian StyleProsdocimi, Francisco, Gabriel S. Zamudio, Miryam Palacios-Pérez, Sávio Torres de Farias, and Marco V. José. 2020. "The Ancient History of Peptidyl Transferase Center Formation as Told by Conservation and Information Analyses" Life 10, no. 8: 134. https://doi.org/10.3390/life10080134
APA StyleProsdocimi, F., Zamudio, G. S., Palacios-Pérez, M., Torres de Farias, S., & V. José, M. (2020). The Ancient History of Peptidyl Transferase Center Formation as Told by Conservation and Information Analyses. Life, 10(8), 134. https://doi.org/10.3390/life10080134