Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients

 ,
, 
 , ,
, , 
Abstract
1. Introduction
2. Material and Methods
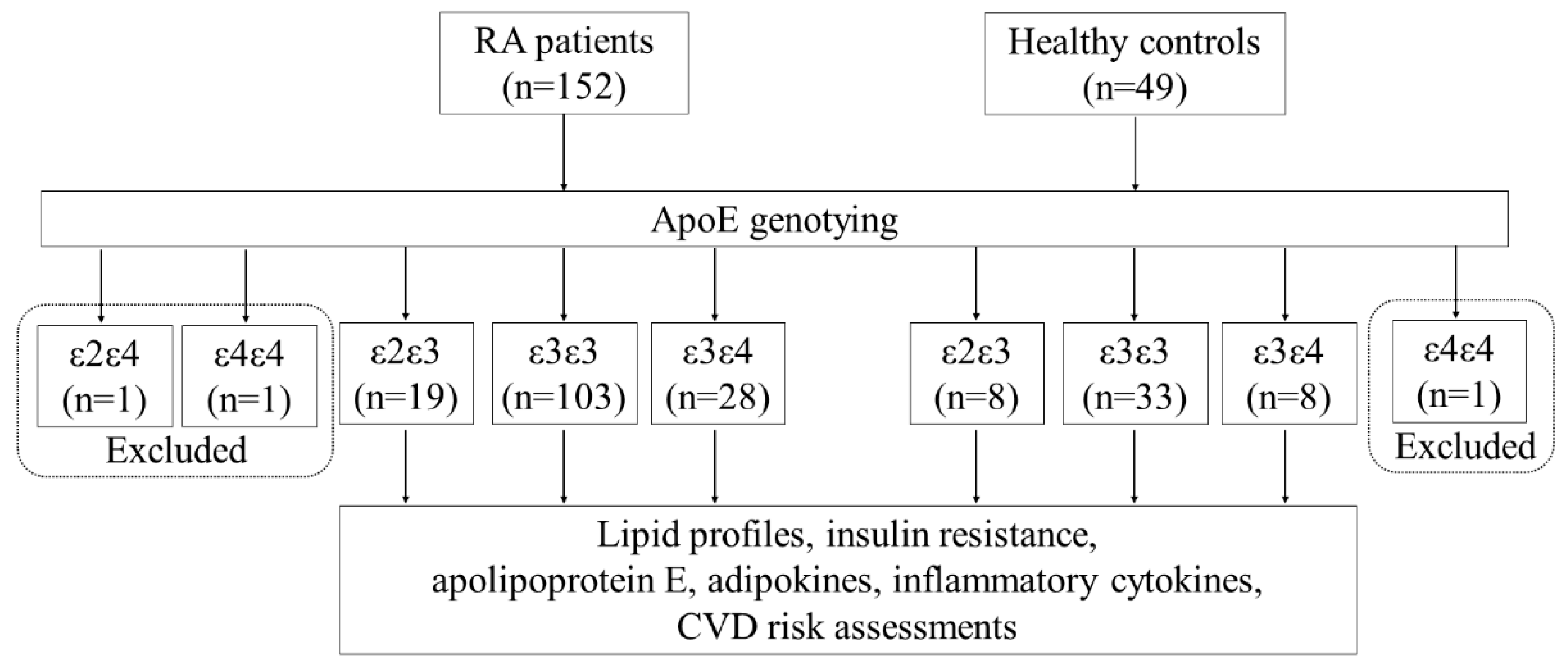
2.1. Subjects
2.2. Determination of Plasma Lipid Profiles and Atherogenic Index (AI)
2.3. Measurements of Insulin Resistance
2.4. Measurement of 10-Year Risk of CVD Including QRISK-2 Score
2.5. Determination of ApoE Genotypes
2.6. Detection of Plasma Levels of ApoE, Adipokines, and Proinflammatory Cytokines
2.7. Statistical Analysis
3. Results
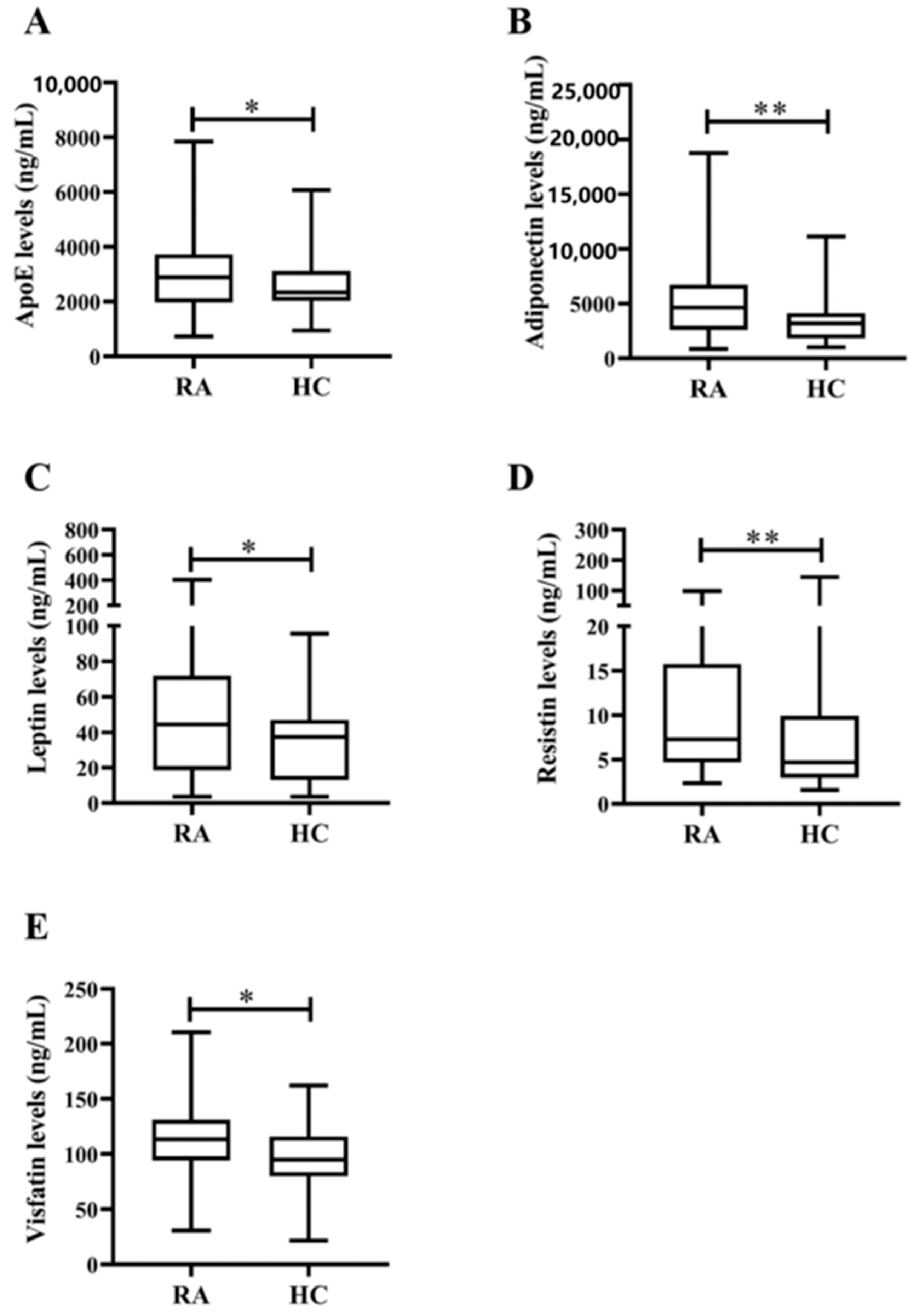
3.1. Demographic Data and Laboratory Findings in RA Patients and Healthy Control Subjects
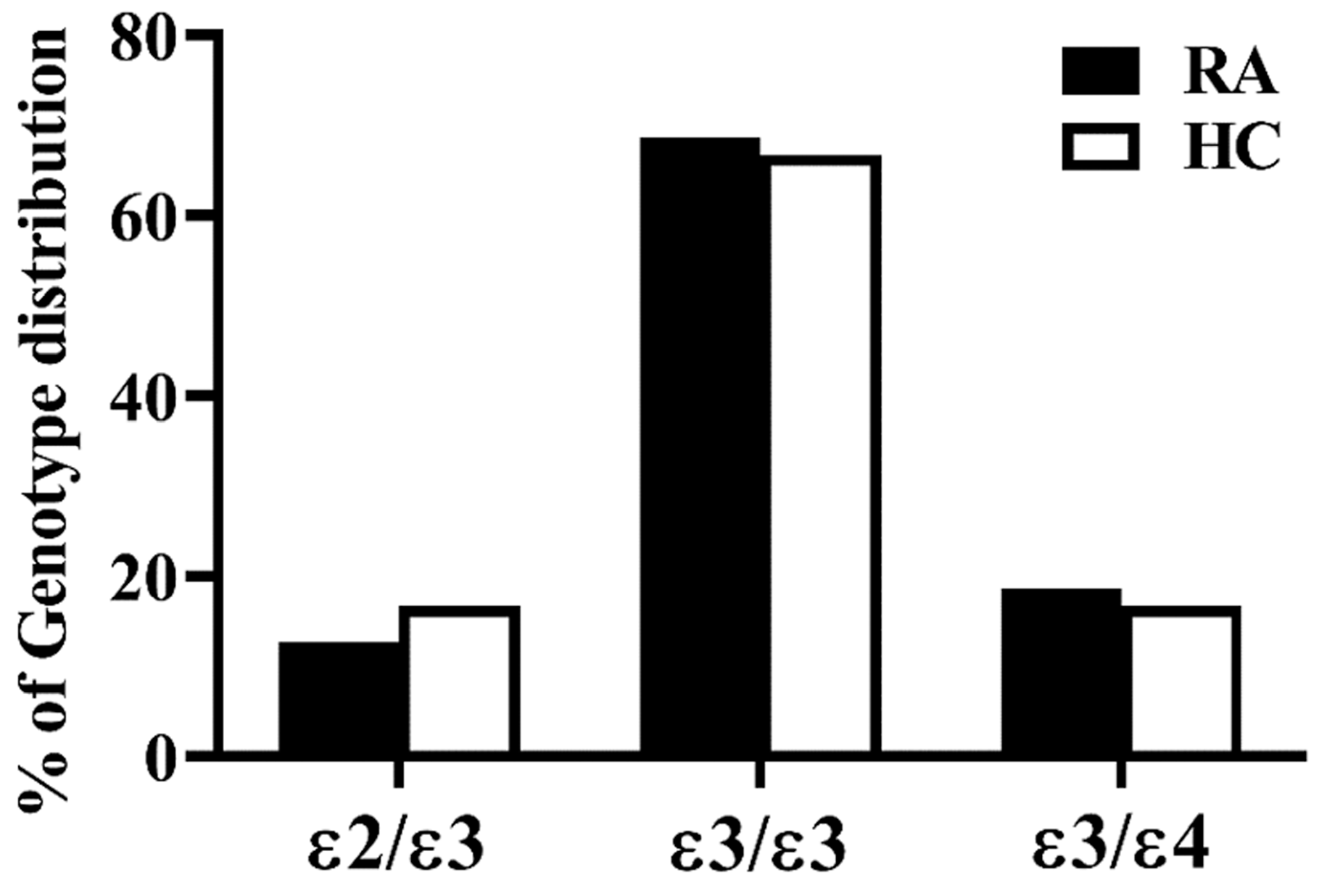
3.2. Frequencies and Distribution of ApoE Genotypes in RA Patients and Healthy Controls
3.3. Clinical Characteristics and Laboratory Findings in RA Patients with Different ApoE Genotypes
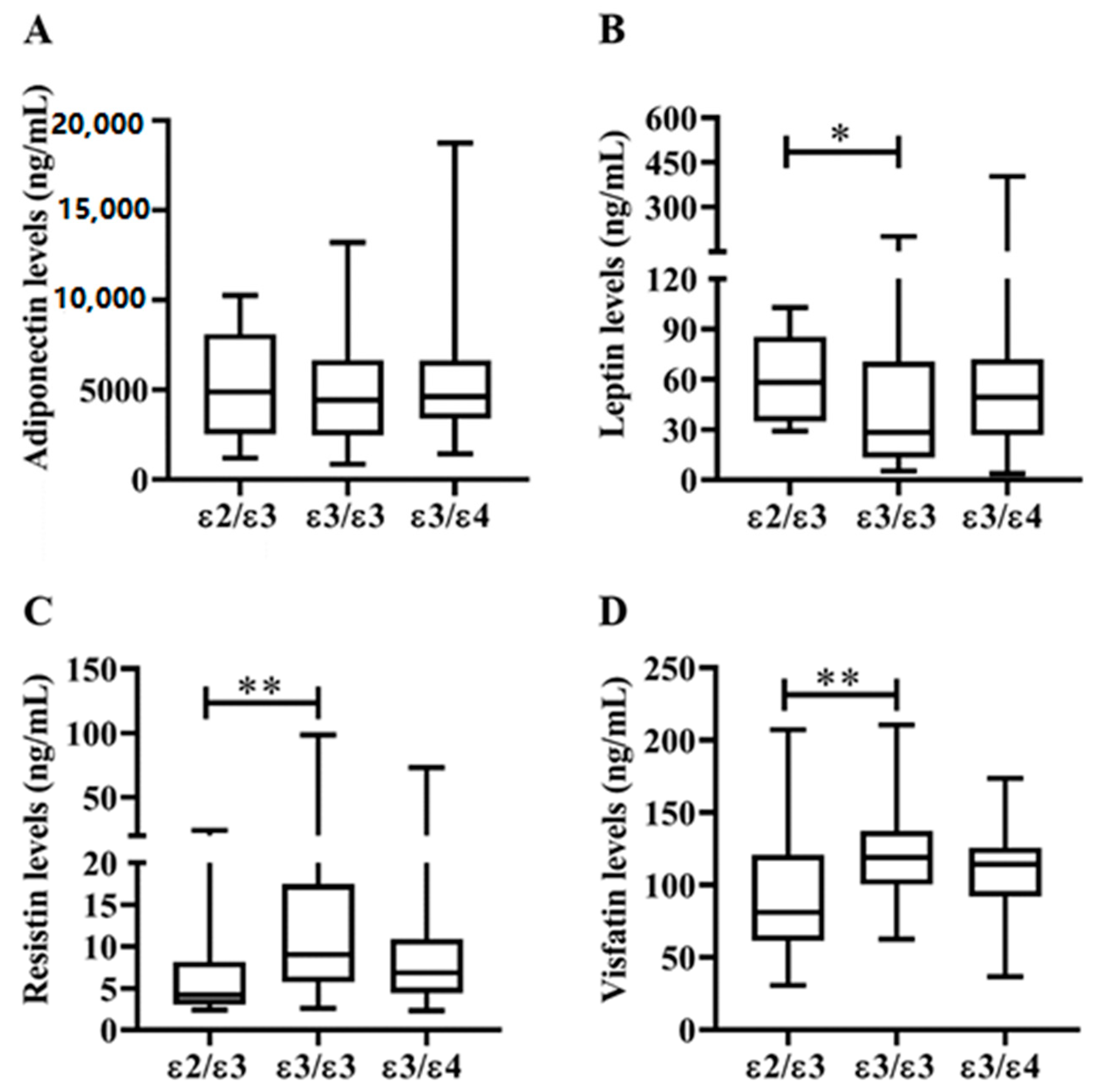
3.4. Correlations between Plasma Adipokines Levels and Inflammatory Parameters or Lipid Profiles in RA Patients with Different ApoE Genotypes
3.5. Logistic Regression Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Libby, P. Inflammation in atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Panayi, G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 2001, 344, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Avina-Zubieta, J.A.; Thomas, J.; Sadatsafavi, M.; Lehman, A.J.; Lacaille, D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2012, 71, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Symmons, D.P.; Gabriel, S.E. Epidemiology of CVD in rheumatic disease, with a focus on RA and SLE. Nat. Rev. Rheumatol. 2011, 7, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Cipriani, P.; Liakouli, V.; Iacono, D.; Pantano, I.; Margiotta, D.P.E.; Navarini, L.; Castaniti, G.M.D.; Maruotti, N.; di Scala, G.; et al. Subclinical and clinical atherosclerosis in rheumatoid arthritis: Results from the 3-year, multicenter, prospective, observational GIRRCS (Gruppo Italiano Di Ricerca in Rheumatologia Clinica E Sperimentale) study. Arthritis. Res. 2019, 21, 204. [Google Scholar] [CrossRef]
- Choy, E.; Ganeshalingam, K.; Semb, A.G.; Iacono, D.; Pantano, I.; Margiotta, D.; Navarini, L.; Destro Castaniti, G.M.; Maruotti, N.; Di Scala, G.; et al. Cardiovascular risk in rheumatoid arthritis: Recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology 2014, 53, 2143–2154. [Google Scholar] [CrossRef]
- Im, C.H.; Kim, N.R.; Kang, J.W.; Kim, J.H.; Kang, J.Y.; Bae, G.B.; Nam, E.J.; Kang, Y.M. Inflammatory burden interacts with conventional cardiovascular risk factors for carotid plaque formation in rheumatoid arthritis. Rheumatology 2015, 54, 808–815. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat. Med. 2003, 9, 1245–1250. [Google Scholar] [CrossRef]
- Furst, D.E.; Emery, P. Rheumatoid arthritis pathophysiology: Update on emerging cytokine and cytokine-associated cell targets. Rheumatology 2014, 53, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Rho, Y.H.; Chung, C.P.; Oeser, A.; Solus, J.; Asanuma, Y.; Sokka, T.; Pincus, T.; Raggi, P.; Gebretsadik, T.; Shintani, A.; et al. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis. Rheum. 2009, 61, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Haddy, N.; Sass, C.; Droesch, S.; Zaiou, M.; Siest, G.; Ponthieux, A.; Lambert, D.; Visvikis, S. IL-6, TNF-α and atherosclerosis risk indicators in healthy family population: The STANISLAS cohort. Atheroslcerosis 2003, 170, 277–283. [Google Scholar] [CrossRef]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Toussirot, E.; Streit, G.; Wendling, D. The contribution of adipose tissue and adipokines to inflammation in joint diseases. Curr. Med. Chem. 2007, 14, 1095–1100. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Gómez, R.; Lago, F.; Dieguez, C.; Gómez-Reino, J.J.; Gualillo, O. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1198–1201. [Google Scholar] [CrossRef]
- Kang, Y.; Park, H.J.; Kang, M.I.; Lee, H.S.; Lee, S.W.; Lee, S.K.; Park, Y.B. Adipokines, inflammation, insulin resistance, and carotid atherosclerosis in patients with rheumatoid arthritis. Arthritis. Res. 2013, 15, R194. [Google Scholar] [CrossRef]
- Larkin, L.; Khachigian, L.M.; Jessup, W. Regulation of apolipoprotein E production in macrophages [Review]. Int. J. Mol. Med. 2000, 6, 253–258. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.M.; Wu, J. Cross-talk between apolipoprotein E and cytokines. Mediat. Inflamm. 2011, 2011, 949072. [Google Scholar] [CrossRef] [PubMed]
- Das, H.K.; McPherson, J.; Bruns, G.A.P.; Karathanasis, S.K.; Breslow, J.L. Isolation, characterization and mapping to chromosome 19 of the human apolipoprotein E gene. J. Biol. Chem. 1985, 260, 6240–6247. [Google Scholar] [PubMed]
- Sing, C.F.; Davignon, J. Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am. J. Hum. Genet. 1985, 37, 268–285. [Google Scholar] [PubMed]
- Van Bockxmeer, F.M.; Mamotte, C.D.S. Apohpoprotein E homozygosity in young men with coronary heart disease. Lancet 1993, 340, 879–880. [Google Scholar] [CrossRef]
- Dzimiri, N.; Meyer, B.F.; Hussain, S.S.; Basco, C.; Afrane, B.; Halees, Z. Relevance of Apolipoprotein E Polymorphism for Coronary Artery Disease in the Saudi Population. Arch. Pathol. Lab. Med. 1999, 123, 1241–1245. [Google Scholar] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Prevoo, M.L.; van’t Hof, M.A.; Kuper, H.H.; van Leeuwen, M.A.; van de Putte, L.B.; van Riel, P.L. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38, 44–48. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Vinogradova, Y.; Robson, J.; Minhas, R.; Sheikh, A.; Brindle, P. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ 2008, 336, 1475–1482. [Google Scholar] [CrossRef]
- QRISK. Available online: https://www.qrisk.org (accessed on 6 December 2020).
- Koch, W.; Ehrenhaft, A.; Griesser, K.; Pfeufer, A.; Müller, J.; Schömig, A.; Kastrati, A. TaqMan systems for genotyping of disease-related polymorphisms present in the gene encoding apolipoprotein E. Clin. Chem. Lab. Med. 2002, 40, 1123–1231. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Aslani, S.; Fadaei, R.; Jamshidi, A.R. New insights to the mechanisms underlying atherosclerosis in rheumatoid arthritis. Int. J. Rheum. Dis. 2017, 20, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Maehlen, M.T.; Provan, S.A.; de Rooy, D.; van der Helm-van Mil, A.H.; Krabben, A.; Saxne, T.; Lindqvist, E.; Semb, A.G.; Uhlig, T.; van der Heijde, D.; et al. Associations between APOE genotypes and disease susceptibility, joint damage and lipid levels in patients with rheumatoid arthritis. PLoS ONE 2013, 8, e60970. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.L.; Kapur, S.; Chinmayi, S.; Fatima, Q.; Pise, H.; Khan, A.; Liyakat, A.; Jilova, V.; Dube, U. A study of genetic markers in patients of rheumatoid arthritis and their co-relation with severity of the disease. J. Assoc. Physicians India 2017, 65, 32–36. [Google Scholar] [PubMed]
- Van Vliet, P.; Mooijaart, S.P.; de Craen, A.J.M.; Rensen, P.C.; van Heemst, D.; Westendorp, R.G. Plasma levels of apolipoprotein E and risk of stroke in old age. Ann. N. Y. Acad. Sci. 2007, 1100, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Boisvert, W.A. Apolipoprotein E and atherosclerosis. Curr. Opin. Lipidol. 2000, 11, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.F.; Gourraud, P.A.; Cantagrel, A.; Davignon, J.L.; Constantin, A. Traditional cardiovascular risk factors in rheumatoid arthritis: A meta-analysis. Jt. Bone Spine 2011, 78, 179–183. [Google Scholar] [CrossRef]
- Toms, T.E.; Smith, J.P.; Panoulas, V.F.; Blackmore, H.; Douglas, K.M.; Kitas, G.D. Apolipoprotein E Gene Polymorphisms Are Strong Predictors of Inflammation and Dyslipidemia in Rheumatoid Arthritis. J. Rheumatol. 2012, 39, 218–225. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di, A.E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef]
- Lehtinen, S.; Lehtimaki, T.; Sisto, T.; Salenius, J.P.; Nikkilä, M.; Jokela, H.; Koivula, T.; Ebeling, F.; Ehnholm, C. Apolipoprotein E polymorphism, serum lipids, myocardial infarction and severity of angiographically verified coronary artery disease in men and women. Atherosclerosis 1995, 114, 83–91. [Google Scholar] [CrossRef]
- Scuteri, A.; Bos, A.J.G.; Zonderman, A.B.; Brant, L.J.; Lakatta, E.G.; Fleg, J.L. Is the apoE4 allele an independent predictor of coronary events? Am. J. Med. 2001, 110, 28–32. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Peasey, A.; Pikhart, H.; Stavek, P.; Kubinova, R.; Marmot, M.; Bobak, M. APOE polymorphism and its effect on plasma C-reactive protein levels in a large general population sample. Hum. Immunol. 2010, 71, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Carrión, M.; Frommer, K.W.; Pérez-García, S.; Müller-Ladner, U.; Gomariz, R.P.; Neumann, E. The adipokine network in rheumatic joint diseases. Int. J. Mol. Sci. 2019, 20, 4091. [Google Scholar]
- Ruscitti, P.; Di Benedetto, P.; Berardicurti, O.; Liakouli, V.; Carubbi, F.; Cipriani, P.; Giacomelli, R. Adipokines in rheumatoid arthritis: The hidden link between inflammation and cardiometabolic comorbidities. J. Immunol. Res. 2018, 2018, 8410182. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Matsuda, M.; Shimomura, I.; Sata, M.; Arita, Y.; Nishida, M.; Maeda, N.; Kumada, M.; Okamoto, Y.; Nagaretani, H.; Nishizawa, H.; et al. Role of adiponectin in preventing vascular stenosis: The missing link of adipo-vascular axis. J. Biol. Chem. 2002, 277, 37487–37491. [Google Scholar] [CrossRef]
- Yadav, A.; Jyoti, P.; Jain, S.K.; Bhattacharjee, J. Correlation of adiponectin and leptin with insulin resistance: A pilot study in healthy north Indian population. Indian. J. Clin. Biochem. 2011, 26, 193–196. [Google Scholar] [CrossRef]
- Kushiyoma, A.; Sakoda, H.; Oue, N.; Okubo, M.; Nakatsu, Y.; Ono, H.; Fukushima, T.; Kamata, H.; Nishimura, F.; Kikuchi, T.; et al. Resistin-like molecule beta is abundantly expressed in foam cells and its involved in atherosclerosis development. Arter. Thromb. Vasc. Biol. 2013, 33, 1986–1993. [Google Scholar] [CrossRef]
- Abeywardena, M.Y.; Leifert, W.R.; Warnes, K.E.; Varghese, J.N.; Head, R.J. Cardiovascular biology of interleukin-6. Curr. Pharm. Des. 2009, 15, 1809–1821. [Google Scholar] [CrossRef][Green Version]
- Sethi, J.K.; Vidal, P.A. Visfatin: The missing link between intra-abdominal obesity and diabetes? Trends. Mol. Med. 2005, 11, 344–347. [Google Scholar] [CrossRef]
- Robinson, C.; Tsang, L.; Solomon, A.; Woodiwiss, A.J.; Gunter, S.; Mer, M.; Hsu, H.C.; Gomes, M.; Norton, G.R.; Millen, A.; et al. Nesfatin-1 and visfatin expression is associated with reduced atherosclerotic disease risk in patients with rheumatoid arthritis. Peptides 2018, 102, 31–37. [Google Scholar] [CrossRef]
- Gibas-Dorna, M.; Piątek, J.; Kupsz, J.; Bernatek, M.; Krauss, H.; Sowińska, A.; Kołodziejski, P.; Owoc, A.; Bojar, I. Relationship between adipokines and lipid profile in postmenopausal women with different apolipoprotein E genotypes. Women Health 2017, 57, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.M.; Frommer, K.W.; Peters, M.A.; Brentano, F.; Lefèvre, S.; Schröder, D.; Kyburz, D.; Steinmeyer, J.; Rehart, S.; Gay, S.; et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J. Biol. Chem. 2012, 287, 28378–28385. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Davidson, S.M.; Paramanathan, A.J.; Smith, C.C.; Yellon, D.M.; Hausenloy, D.J. The novel adipocytokine visfatin exerts direct cardioprotective effects. J. Cell. Mol. Med. 2008, 12, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Stampfer, M.J.; Liu, S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann. Intern. Med. 2004, 141, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.D.; Gu, W.; Qiao, S.B.; Zhu, E.J.; Zhao, Q.M.; Lv, S.Z. Apolipoprotein E polymorphism and risk for coronary heart disease in the Chinese population: A meta-analysis of 61 studies including 6634 cases and 6393 controls. PLoS ONE 2014, 9, e95463. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.L.; Edwards, C.J. Protective effect of methotrexate in patients with rheumatoid arthritis and cardiovascular comorbidity. Adv. Musculoskel. Dis. 2012, 4, 149–157. [Google Scholar] [CrossRef]
- Burska, A.N.; Sakthiswary, R.; Sattar, N. Effects of Tumour Necrosis Factor Antagonists on Insulin Sensitivity/Resistance in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0128889. [Google Scholar] [CrossRef]
- Rempenault, C.; Combe, B.; Barnetche, T.; Gaujoux-Viala, C.; Lukas, C.; Morel, J.; Hua, C. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2018, 77, 98–103. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RA (n = 150) | HC (n = 48) | |
|---|---|---|
| Mean age (years) | 52.8 ± 11.6 | 51.4 ± 7.3 |
| Female (%) | 123 (82.0%) | 36 (75.0%) |
| Disease duration (years) | 15.5 ± 10.2 | - |
| DAS-28 at study entry | 4.85 ± 1.27 | - |
| Smoking (ever) (%) | 30 (20.0%) | 6 (12.5%) |
| Apolipoprotein E, ng/mL | 2885.5 (1967.2–3731.2) * | 2341.3 (2027.4–3117.9) |
| Adiponectin, levels, ng/mL | 4629.8 (2593.9–6714.7) ** | 3191.1 (1817.0–4143.2) |
| Leptin levels, ng/mL | 44.4 (18.6–71.8) * | 37.3 (13.1–47.1) |
| Resistin levels, ng/mL | 7.3 (4.7–15.7) ** | 4.7 (2.9–9.9) |
| Visfatin levels, ng/mL | 113.4 (94.3–131.2) * | 94.8 (79.9–116.0) |
| TNF-α levels, pg/mL | 31.9 (12.7–56.4) | 28.1 (10.0–36.5) |
| Interleukin-6 levels, pg/mL | 101.2 (56.3–389.0) | 96.9 (53.5–205.5) |
| Interleukin-17A levels, pg/mL | 110.2 (63.9–176.4) ** | 68.2 (36.1–114.8) |
| Daily steroid dose, mg | 6.4 ± 2.6 | - |
| csDMARDs alone | 99 (66.0%) | |
| Methotrexate dose, mg per week | 9.0 ± 5.7 | - |
| TNF inhibitors | 33 (22.0%) | - |
| Rituximab | 7 (4.7%) | - |
| Tocilizumab | 11 (7.3%) | - |
| ε2ε3 (n = 19) | ε3ε3 (n = 103) | ε3ε4 (n = 28) | |
|---|---|---|---|
| Mean age (years) | 56.7 ± 12.1 | 55.5 ± 14.1 | 58.1 ± 14.3 |
| Female (%) | 17 (89.5%) | 83 (80.6%) | 23 (82.1%) |
| Disease duration (years) | 15.3 ± 5.0 | 14.4 ± 6.1 | 13.4 ± 7.8 |
| RF positivity (%) | 11 (57.9%) | 77 (74.8%) | 19 (67.9%) |
| ACPA positivity (%) | 13 (68.4%) | 70 (68.0%) | 18 (64.3%) |
| Smoking (ever) (%) | 2 (10.5%) | 22 (21.4%) | 6 (21.4%) |
| HT (%) | 8 (42.1%) | 53 (51.5%) | 14 (50.0%) |
| DM (%) | 2 (10.5%) | 17 (16.5%) | 6 (21.4%) |
| Body mass index, kg/m2 | 24.0 ± 3.03 | 24.1 ± 3.01 | 23.5 ± 2.33 |
| Insulin resistance (HOMA) | 1.83 ± 1.25 | 2.55 ± 2.90 | 4.05 ± 7.32 |
| CVD event (%) | 1 (5.3%) | 11 (10.7%) | 5 (17.9%) |
| Qrisk-2 score | 4.25 ± 3.45 * | 7.28 ± 8.49 | 8.76 ± 11.2 |
| Total cholesterol, mg/dL | 185 (159–214) | 198 (173–227) | 185 (172–217) |
| HDL-C, mg/dL | 72 (60–80) | 67 (57–88) | 59 (53–74) |
| LDL-C, mg/dL | 95 (89–124) # | 120 (96–146) | 122 (103–141) |
| Atherogenic index | 2.44 (2.27–2.76) # | 2.84 (2.36–3.43) | 3.06 (2.77–3.63) |
| Triglyceride, mg/dL | 83 (66–133) | 96 (67–122) | 83 (67–126) |
| TNF-α levels, pg/mL | 9.4 (6.8–45.9) * | 36.2 (24.4–66.0) | 30.4 (8.9–48.8) |
| IL-6 levels, pg/mL | 60.9 (53.6–101.2) * | 115.1 (60.3–505.4) | 93.1 (44.4–224.0) |
| IL-17A levels, pg/mL | 109.3 (56.3–124.6) | 116.6 (65.0–241.8) | 87.4 (58.1–132.2) |
| CRP, at entry, mg/dl | 0.42 ± 0.54 # | 1.24 ± 2.72 | 1.40 ± 1.45 * |
| DAS-28 at study entry | 3.95 ± 1.34 | 4.19 ± 1.39 | 4.65 ± 1.36 |
| Apolipoprotein E, ng/mL | 3622 (3163–5433) *## | 3000 (2167–3931) | 1913 (1466–2735) *** |
| Adiponectin, levels, ng/mL | 4870 (2520–8104) | 4417 (2465–6650) | 4630 (3391–6640) |
| Leptin levels, ng/mL | 58.3 (34.9–85.5) ** | 28.3 (13.2–70.5) | 49.2 (26.8–72.0) |
| Resistin levels, ng/mL | 4.2 (3.0–8.2) ** | 9.0 (5.7–17.4) | 6.9 (4.4–10.9) |
| Visfatin levels, ng/mL | 81.1 (61.6–120.7) ** | 119.0 (100.4–137.4) | 114.3 (92.1–125.7) |
| Daily steroid dose, mg | 3.4 ± 2.5 # | 4.2 ± 2.7 | 5.1 ± 2.8 |
| The used DMARDs at entry | |||
| csDMARDs alone | 14 (73.7%) | 67 (65.0%) | 18 (64.3%) |
| TNF inhibitors | 2 (10.5%) | 25 (24.3%) | 6 (21.4%) |
| Rituximab | 1 (5.3%) | 5 (4.9%) | 1 (3.6%) |
| Tocilizumab | 2 (10.5%) | 6 (5.8%) | 3 (10.7%) |
| ApoE ε2ε3 Genotype (n = 19) | Adiponectin | Leptin | Resistin | Visfatin |
|---|---|---|---|---|
| Disease duration | 0.469 | 0.029 | 0.374 | −0.060 |
| CRP levels | 0.433 | 0.345 | 0.391 | 0.352 |
| TNF-α levels | 0.345 | 0.552 * | 0.427 | 0.421 |
| IL-6 levels | −0.130 | 0.252 | 0.527 * | −0.118 |
| IL-17A levels | −0.140 | 0.527 * | 0.176 | −0.056 |
| Total cholesterol levels | 0.053 | 0.280 | −0.215 | 0.321 |
| Triglyceride levels | 0.044 | 0.678 ** | −0.255 | 0.159 |
| HDL-C levels | 0.181 | −0.139 | 0.016 | −0.280 |
| LDL-C levels | 0.202 | 0.375 | −0.195 | 0.504 |
| Atherogenic index | −0.182 | 0.469 | −0.047 | 0.509 * |
| Insulin resistance (HOMA) | −0.181 | 0.657 * | −0.044 | 0.220 |
| Body mass index | −0.059 | 0.074 | −0.315 | 0.048 |
| ApoE ε3ε3 Genotype (n = 103) | Adiponectin | Leptin | Resistin | Visfatin |
| Disease duration | 0.145 | 0.091 | 0.177 | 0.086 |
| CRP levels | −0.242 | −0.140 | −0.153 | 0.173 |
| TNF-α levels | −0.061 | 0.062 | 0.075 | 0.238 |
| IL-6 levels | 0.100 | 0.216 | −0.115 | 0.237 |
| IL-17A levels | 0.326 * | 0.245 | −0.153 | 0.105 |
| Total cholesterol levels | 0.280 | 0.149 | 0.137 | 0.014 |
| Triglyceride levels | −0.139 | −0.042 | −0.255 | 0.272 |
| HDL-C levels | 0.297 * | 0.251 | 0.177 | −0.174 |
| LDL-C levels | 0.171 | −0.028 | 0.136 | 0.098 |
| Atherogenic index | −0.093 | −0.204 | 0.020 | 0.149 |
| Insulin resistance (HOMA) | −0.395 * | −0.114 | −0.097 | 0.349 * |
| Body mass index | −0.036 | −0.024 | −0.044 | 0.302 * |
| ApoE ε3ε4 Genotype (n = 28) | Adiponectin | Leptin | Resistin | Visfatin |
| Disease duration | 0.287 | −0.175 | 0.227 | 0.317 |
| CRP levels | −0.014 | 0.284 | 0.090 | −0.032 |
| TNF-α levels | −0.044 | 0.886 ** | −0.334 | 0.096 |
| IL-6 levels | 0.186 | 0.325 | −0.138 | −0.343 |
| IL-17A levels | 0.213 | 0.064 | 0.076 | 0.234 |
| Total cholesterol levels | 0.244 | −0.193 | 0.052 | 0.290 |
| Triglyceride levels | −0.003 | −0.062 | 0.192 | 0.110 |
| HDL-C levels | 0.366 | −0.111 | 0.141 | 0.433 |
| LDL-C levels | 0.145 | −0.002 | −0.065 | 0.109 |
| Atherogenic index | −0.132 | 0.006 | −0.074 | −0.318 |
| Insulin resistance (HOMA) | −0.311 | 0.020 | 0.560 * | 0.325 |
| Body mass index | −0.270 | 0.227 | 0.355 | 0.041 |
| Risk Factors (Univariate) | β-Value | 95% Confidence Interval | p-Value |
|---|---|---|---|
| Constant | |||
| ApoE genotype | |||
| ε2ε3 | Reference | Reference | |
| ε3ε3 | 3.03 | (−1.22, 7.28) | 0.162 |
| ε3ε4 | 4.51 | (−0.55, 9.58) | 0.080 |
| ACPA | |||
| Negativity | Reference | Reference | |
| Positivity | 3.58 | (0.65, 6.51) | 0.017 |
| TNF-α levels, pg/mL | −0.0010 | (−0.0033, 0.0024) | 0.755 |
| IL-6 levels, pg/mL | 0.0003 | (−0.0006, 0.0012) | 0.495 |
| IL-17A levels, pg/mL | −0.0005 | (−0.0055, 0.0044) | 0.828 |
| Adiponectin, levels, ng/mL | 0.0006 | (0.0002, 0.0010) | 0.007 |
| Leptin levels, ng/mL | 0.005 | (−0.021, 0.030) | 0.722 |
| Resistin levels, ng/mL | 0.04 | (−0.05, 0.12) | 0.403 |
| Visfatin levels, ng/mL | 0.02 | (−0.02, 0.07) | 0.229 |
| Atherogenic index | 3.06 | (1.52, 4.60) | <0.001 |
| Risk Factors (Multivariate) | β-Value | 95% Confidence Interval | p-Value |
| ApoE genotype | |||
| ε3ε4 | 4.86 | (0.62, 9.10) | 0.025 |
| ACPA | |||
| Positivity | 3.17 | (0.08, 6.25) | 0.045 |
| Adiponectin, levels, ng/mL | 0.0005 | (0.0001, 0.0010) | 0.012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-M.; Chen, P.-K.; Chang, C.-K.; Lin, C.-C.; Chen, H.-H.; Lan, J.-L.; Chang, S.-H.; Chen, D.-Y. Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients. Life 2020, 10, 330. https://doi.org/10.3390/life10120330
Chen Y-M, Chen P-K, Chang C-K, Lin C-C, Chen H-H, Lan J-L, Chang S-H, Chen D-Y. Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients. Life. 2020; 10(12):330. https://doi.org/10.3390/life10120330
Chicago/Turabian StyleChen, Yi-Ming, Po-Ku Chen, Ching-Kun Chang, Chi-Chen Lin, Hsin-Hua Chen, Joung-Liang Lan, Shih-Hsin Chang, and Der-Yuan Chen. 2020. "Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients" Life 10, no. 12: 330. https://doi.org/10.3390/life10120330
APA StyleChen, Y.-M., Chen, P.-K., Chang, C.-K., Lin, C.-C., Chen, H.-H., Lan, J.-L., Chang, S.-H., & Chen, D.-Y. (2020). Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients. Life, 10(12), 330. https://doi.org/10.3390/life10120330