Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites and Sample Collection
2.2. Plasmodium Diagnosis by Microscopy and PCR
2.3. Plasmodium falciparum Genotyping
2.4. Data and Statistical Analysis
2.5. Ethical Statement
3. Results
3.1. Allelic Frequency of glurp, msp-1 and msp-2
3.2. Haplotype Diversity and Distribution
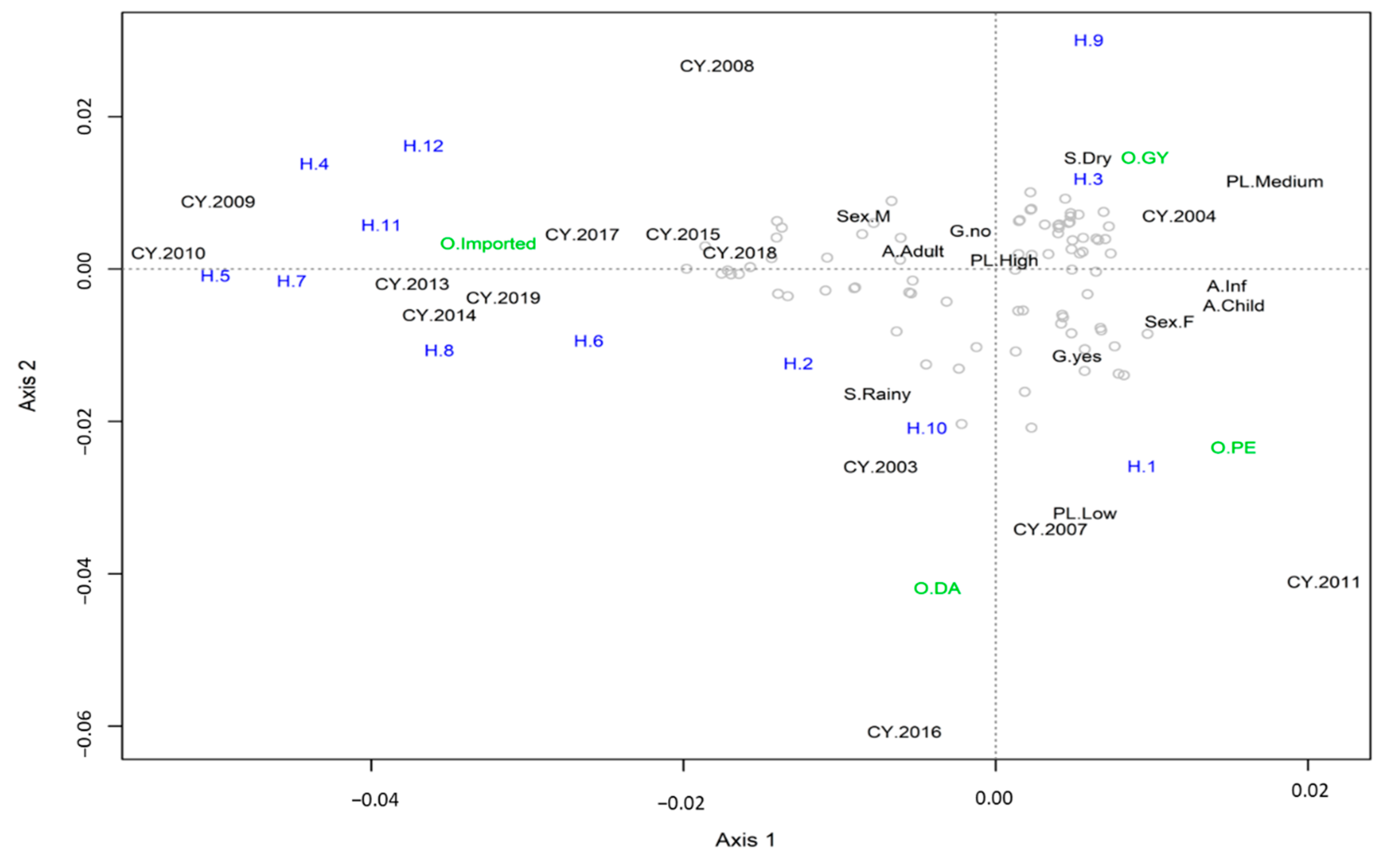
3.3. Multiple Correspondence Analysis
3.4. Msp-2 Sequencing and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Artemisinin-based combination therapy |
| CQ | Chloroquine |
| DCV-MoH | Departamento de Control de Vectores from the Ministry of Health |
| glurp | Glutamate-rich protein |
| ICGES | Instituto Conmemorativo Gorgas de Estudios de la Salud |
| MCA | Multiple correspondence analysis |
| msp-1 | Merozoite surface proteins 1 |
| msp-2 | Merozoite surface proteins 2 |
| MOI | Multiplicity of infection |
| NMEP | National Malaria Elimination Programme |
| RFLP | Restriction fragment length polymorphism |
| SP | Sulfadoxine–pyrimethamine |
References
- Ministerio de Salud. Plan Estratégico de Eliminación de la Malaria (PEEM) en Panamá 2018–2022; Ministerio de Salud: República de Panamá, Panamá, 2018; pp. 8–39.
- Obaldia, N., III. Determinants of low socio-economic status and risk of Plasmodium vivax malaria infection in Panama (2009–2012): A case-control study. Malar. J. 2015, 14, 14. [Google Scholar] [CrossRef]
- Hurtado, L.A.; Calzada, J.E.; Rigg, C.A.; Castillo, M.; Chaves, L.F. Climatic fluctuations and malaria transmission dynamics, prior to elimination, in Guna Yala, Republica de Panamá. Malar. J. 2018, 17, 85. [Google Scholar] [CrossRef]
- Rigg, C.A.; Hurtado, L.A.; Calzada, J.E.; Chaves, L.F. Malaria infection rates in Anopheles albimanus (Diptera: Culicidae) at Ipetí-Guna, a village within a region targeted for malaria elimination in Panamá. Infect. Genet. Evol. 2019, 69, 216–223. [Google Scholar] [CrossRef]
- Cáceres-Carrera, L.; Victoria, C.; Ramirez, J.L.; Jackman, C.; Calzada, J.E.; Torres, R. Study of the epidemiological behavior of malaria in the Darien Region, Panama. 2015–2017. PLoS ONE 2019, 14, e0224508. [Google Scholar] [CrossRef]
- Hurtado, L.A.; Rigg, C.A.; Calzada, J.E.; Dutary, S.; Bernal, D.; Koo, S.I.; Chaves, L.F. Population Dynamics of Anopheles albimanus (Diptera: Culicidae) at Ipetí-Guna, a Village in a Region Targeted for Malaria Elimination in Panamá. Insects 2018, 9, 164. [Google Scholar] [CrossRef]
- Hurtado, L.; Cumbrera, A.; Rigg, C.; Perea, M.; Santamaría, A.M.; Chaves, L.F.; Moreno, D.; Romero, L.; Lasso, J.; Caceres, L.; et al. Long-term transmission patterns and public health policies leading to malaria elimination in Panamá. Malar. J. 2020, 19, 265. [Google Scholar] [CrossRef]
- Samudio, F.; Santamaría, A.M.; Obaldía, N., III; Pascale, J.M.; Bayard, V.; Calzada, J.E. Prevalence of Plasmodium falciparum mutations associated with antimalarial drug resistance during an epidemic in Kuna Yala, Panama, Central America. Am. J. Trop. Med. Hyg. 2005, 73, 839–841. [Google Scholar] [CrossRef]
- Calzada, J.E.; Samudio, F.; Bayard, V.; Obaldia, N., III; de Mosca, I.B.; Pascale, J.M. Revising antimalarial drug policy in Central America: Experience in Panama. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 694–698. [Google Scholar] [CrossRef]
- Obaldia, N., III; Baro, N.K.; Calzada, J.E.; Santamaria, A.M.; Daniels, R.; Wong, W.; Chang, H.H.; Hamilton, E.J.; Arevalo-Herrera, M.; Herrera, S.; et al. Clonal outbreak of Plasmodium falciparum infection in eastern Panama. J. Infect. Dis. 2015, 211, 1087–1096. [Google Scholar] [CrossRef]
- Ministerio de Salud Panamá. Manual de Normas y Procedimientos para Malaria; Ministerio de Salud Panamá: República de Panamá, Panamá, 2011; pp. 36–317.
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015; p. 316. [Google Scholar]
- Calzada, J.E.; Marquez, R.; Rigg, C.; Victoria, C.; De La Cruz, M.; Chaves, L.F.; Cáceres, L. Characterization of a recent malaria outbreak in the autonomous indigenous region of Guna Yala, Panama. Malar. J. 2015, 14, 459. [Google Scholar] [CrossRef]
- Herrera, S.; Ochoa-Orozco, S.A.; González, I.J.; Peinado, L.; Quiñones, M.L.; Arévalo-Herrera, M. Prospects for malaria elimination in Mesoamerica and Hispaniola. PLoS Negl. Trop. Dis. 2015, 9, e0003700. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, W.; Dutari, L.C.; Rovira, J.R.; Sucupira, I.M.; Póvoa, M.M.; Conn, J.E.; Loaiza, J.R. Epidemic and Non-Epidemic Hot Spots of Malaria Transmission Occur in Indigenous Comarcas of Panama. PLoS Negl. Trop. Dis. 2016, 10, e0004718. [Google Scholar] [CrossRef] [PubMed]
- Kotepui, M.; Piwkham, D.; PhunPhuech, B.; Phiwklam, N.; Chupeerach, C.; Duangmano, S. Effects of malaria parasite density on blood cell parameters. PLoS ONE 2015, 10, e0121057. [Google Scholar] [CrossRef] [PubMed]
- Snounou, G.; Viriyakosol, S.; Jarra, W.; Thaithong, S.; Brown, K.N. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 1993, 58, 283–292. [Google Scholar] [CrossRef]
- Snewin, V.A.; Herrera, M.; Sanchez, G.; Scherf, A.; Langsley, G.; Herrera, S. Polymorphism of the alleles of the merozoite surface antigens MSA1 and MSA2 in Plasmodium falciparum wild isolates from Colombia. Mol. Biochem. Parasitol. 1991, 49, 265–275. [Google Scholar] [CrossRef]
- Ranford-Cartwright, L.C.; Taylor, J.; Umasunthar, T.; Taylor, L.H.; Babiker, H.A.; Lell, B.; Schmidt-Ott, J.R.; Lehman, L.G.; Walliker, D.; Kremsner, P.G. Molecular analysis of recrudescent parasites in a Plasmodium falciparum drug efficacy trial in Gabon. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 719–724. [Google Scholar] [CrossRef]
- Felger, I.; Irion, A.; Steiger, S.; Beck, H.P. Genotypes of merozoite surface protein 2 of Plasmodium falciparum in Tanzania. Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 3–9. [Google Scholar] [CrossRef]
- Kidima, W.; Nkwengulila, G. Plasmodium falciparum msp2 Genotypes and Multiplicity of Infections among Children under Five Years with Uncomplicated Malaria in Kibaha, Tanzania. J. Parasitol. Res. 2015, 2015, 721201. [Google Scholar] [CrossRef]
- Felger, I.; Beck, H.P. Genotyping of Plasmodium falciparum. In Malaria Methods and Protocols: Methods in Molecular Medicine; Doolan, D.L., Ed.; Humana Press: Totawa, NJ, USA, 2002; Volume 72, pp. 117–129. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Rigg, C.A.; Calzada, J.E.; Saldaña, A.; Perea, M.; Chaves, L.F.; Valderrama, A. Leishmania spp. Infection Rate and Feeding Patterns of Sand Flies (Diptera: Psychodidae) from a Hyperendemic Cutaneous Leishmaniasis Community in Panamá. Am. J. Trop. Med. Hyg. 2019, 100, 798–807. [Google Scholar] [CrossRef]
- Consejo de Ministros de Salud de Centroamérica y República Dominicana (COMISCA). Declaración-Hacia la Eliminación de la Malaria en Mesoamérica y la Isla de la Española en el 2020; COMISCA XROd: San José, Costa Rica, 2013.
- Hurtado, L.A.; Cáceres, L.; Chaves, L.F.; Calzada, J.E. When climate change couples social neglect: Malaria dynamics in Panamá. Emerg. Microbes Infect. 2014, 3, 1–11. [Google Scholar] [CrossRef]
- Cáceres, L.; Calzada, J.E.; Gabster, A.; Young, J.; Márquez, R.; Torres, R.; Griffith, M. Social representations of malaria in the Guna indigenous population of Comarca Guna de Madungandi, Panama. Malar. J. 2017, 16, 256. [Google Scholar] [CrossRef]
- Sturrock, H.J.W.; Roberts, K.W.; Wegbreit, J.; Ohrt, C.; Gosling, R.D. Tackling imported malaria: An elimination endgame. Am. J. Trop. Med. Hyg. 2015, 93, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Yates, C. As More Migrants from Africa and Asia Arrive in Latin America, Governments Seek Orderly and Controlled Pathways. Available online: https://www.migrationpolicy.org/article/extracontinental-migrants-latin-america (accessed on 22 October 2019).
- Ndao, M.; Bandyayera, E.; Kokoskin, E.; Gyorkos, T.W.; MacLean, J.D.; Ward, B.J. Comparison of blood smear, antigen detection, and nested-PCR methods for screening refugees from regions where malaria is endemic after a malaria outbreak in Quebec, Canada. J. Clin. Microbiol. 2004, 42, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Monge-Maillo, B.; López-Vélez, R. Is screening for malaria necessary among asymptomatic refugees and immigrants coming from endemic countries? Expert Rev. Anti-Infect. Ther. 2011, 9, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.; Hall, L. Malaria on the move: Human population movement and malaria transmission. Emerg. Infect. Dis. 2000, 6, 103–109. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. A Framework for Malaria Elimination; World Health Organization: Geneva, Switzerland, 2017; p. 100. [Google Scholar]
- Hotez, P.J.; Woc-Colburn, L.; Bottazzi, M.E. Neglected tropical diseases in Central America and Panama: Review of their prevalence, populations at risk and impact on regional development. Int. J. Parasitol. 2014, 44, 597–603. [Google Scholar] [CrossRef]
- Van Tyne, D.; Park, D.J.; Schaffner, S.F.; Neafsey, D.E.; Angelino, E.; Cortese, J.F.; Barnes, K.G.; Rosen, D.M.; Lukens, A.K.; Daniels, R.F.; et al. Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in Plasmodium falciparum. PLoS Genet. 2011, 7, e1001383. [Google Scholar] [CrossRef]
- Lopez, A.C.; Ortiz, A.; Coello, J.; Sosa-Ochoa, W.; Torres, R.E.M.; Banegas, E.I.; Jovel, I.; Fontecha, G.A. Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malar. J. 2012, 11, 391. [Google Scholar] [CrossRef]
- Medicines for Malaria Venture & World Health Organization. Methods and Techniques for Clinical Trials on Antimalarial Drug Efficacy: Genotyping to Identify Parasite Populations; Informal Consultation Organized by the Medicines for Malaria Venture and Cosponsored; World Health Organization: Amsterdam, The Netherlands, 2007; p. 45. [Google Scholar]
- World Health Organization. Update on the e-2020 Initiative of 21 Malaria-Eliminating Countries: Report and Country Briefs; World Health Organization: Geneva, Switzerland, 2018; p. 51. [Google Scholar]
- Bennett, A.; Smith, J.L. Malaria Elimination: Lessons from El Salvador. Am. J. Trop. Med. Hyg. 2018, 99, 1–2. [Google Scholar] [CrossRef]
- Marín Rodríguez, R.; Chaves, L.F. Parasite Removal for Malaria Elimination in Costa Rica. Trends Parasitol. 2019, 35, 585–588. [Google Scholar] [CrossRef]
- Chaves, L.F.; Huber, J.H.; Rojas Salas, O.; Ramírez Rojas, M.; Romero, L.M.; Gutiérrez Alvarado, J.M.; Perkins, T.A.; Prado, M.; Marín Rodríguez, R. Malaria Elimination in Costa Rica: Changes in Treatment and Mass Drug Administration. Microorganisms 2020, 8, 984. [Google Scholar] [CrossRef] [PubMed]
- Chaves, L.F.; Ramírez Rojas, M.; Prado, M.; Garcés, J.L.; Salas Peraza, D.; Marín Rodríguez, R. Health policy impacts on malaria transmission in Costa Rica. Parasitology 2020, 147, 999–1007. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Gene Alleles | Case Origin | No | Total | ||||
|---|---|---|---|---|---|---|---|---|
| glurp | msp-2 | msp-1 | Indigenous | No | Imported | |||
| 1 | 1100 bp | 3D7 | 500 bp | Panamá Este | 17 | Africa | 3 | 22 |
| Darién | 2 | |||||||
| 2 | 1100 bp | 3D7/FC27 | 500 bp | Panamá Este | 1 | Central Africa | 1 | 2 |
| 3 | 800 bp | 3D7 | 500 bp | Guna Yala | 26 | South America * | 1 | |
| Darién | 3 | Egypt | 1 | |||||
| Panamá Este | 1 | Tanzania | 1 | 35 | ||||
| Zimbabwe | 1 | |||||||
| Africa ** | 1 | |||||||
| 4 | 1100 bp | FC27 | 500 bp | Haiti | 1 | 2 | ||
| Zimbabwe | 1 | |||||||
| 5 | 1000 bp | FC27 | 500 bp | Philippines | 1 | 2 | ||
| India | 1 | |||||||
| 6 | 900 bp | FC27 | 500 bp | China | 1 | 2 | ||
| Africa | 1 | |||||||
| 7 | 1000 bp | 3D7 | 500 bp | Colombia | 1 | 3 | ||
| Nigeria | 1 | |||||||
| Equatorial Guinea | 1 | |||||||
| 8 | 1100 bp | 3D7 | 500 bp | West Africa | 1 | 1 | ||
| 9 | 800 bp | 3D7/FC27 | 500 bp | Guna Yala | 1 | 1 | ||
| 10 | 800 bp | FC27 | 500 bp | Darién | 2 | 4 | ||
| Guna Yala | 2 | |||||||
| 11 | 1000 bp | 3D7 | 600 bp | Cameroon | 2 | 3 | ||
| India | 1 | |||||||
| 12 | 1100 bp | FC27 | 600 bp | Cameroon | 1 | 1 | ||
| Total | 55 | 23 | 78 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santamaría, A.M.; Vásquez, V.; Rigg, C.; Moreno, D.; Romero, L.; Justo, C.; Chaves, L.F.; Saldaña, A.; Calzada, J.E. Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica. Life 2020, 10, 319. https://doi.org/10.3390/life10120319
Santamaría AM, Vásquez V, Rigg C, Moreno D, Romero L, Justo C, Chaves LF, Saldaña A, Calzada JE. Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica. Life. 2020; 10(12):319. https://doi.org/10.3390/life10120319
Chicago/Turabian StyleSantamaría, Ana María, Vanessa Vásquez, Chystrie Rigg, Dianik Moreno, Luis Romero, Carlos Justo, Luis Fernando Chaves, Azael Saldaña, and José E. Calzada. 2020. "Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica" Life 10, no. 12: 319. https://doi.org/10.3390/life10120319
APA StyleSantamaría, A. M., Vásquez, V., Rigg, C., Moreno, D., Romero, L., Justo, C., Chaves, L. F., Saldaña, A., & Calzada, J. E. (2020). Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica. Life, 10(12), 319. https://doi.org/10.3390/life10120319