Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections

 , , ,
, , , 
Abstract
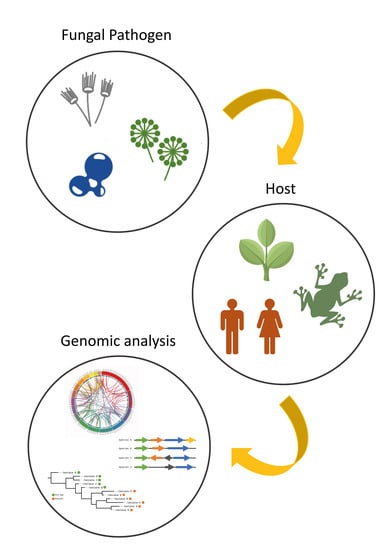
{kind=link}
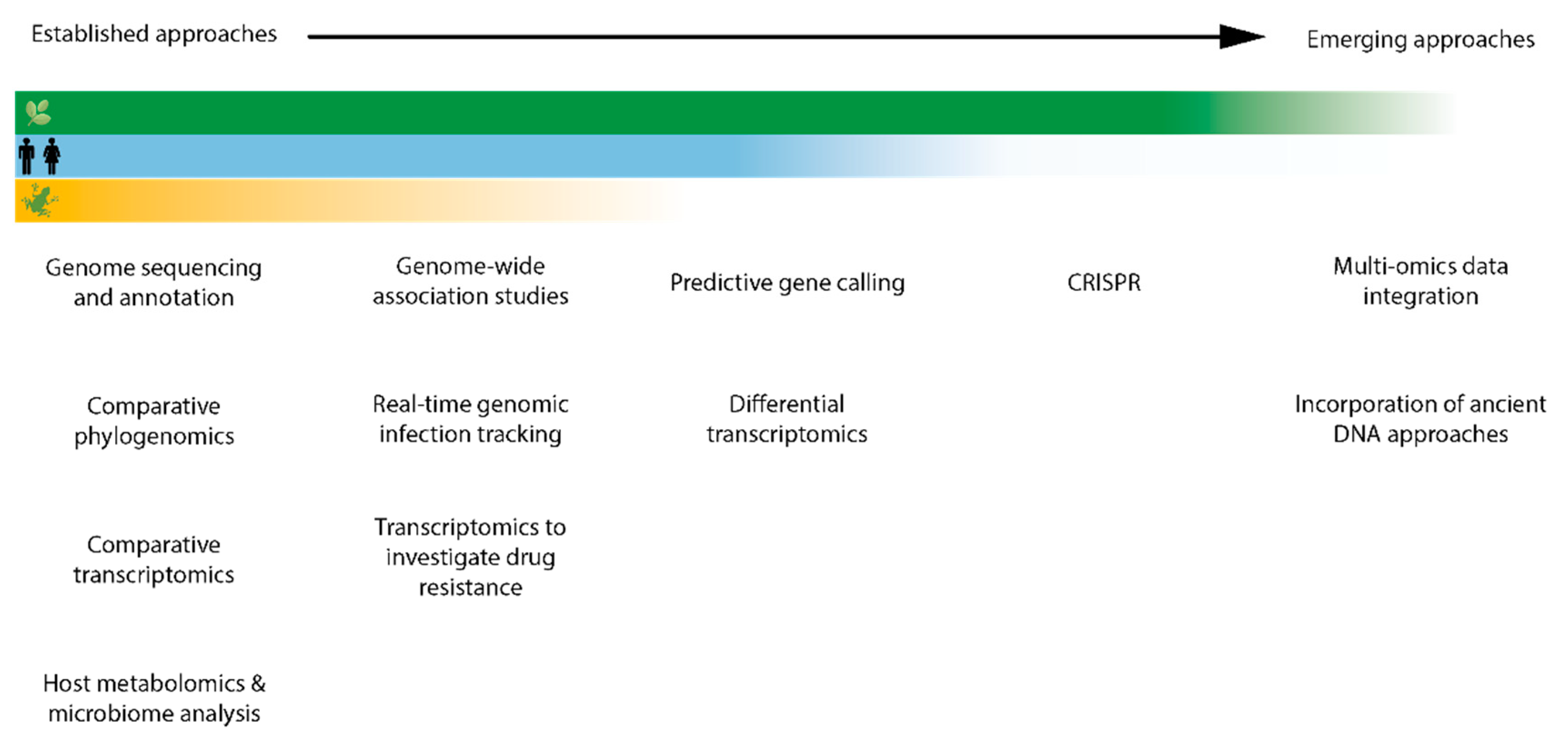{kind=link}
1. Introduction
2. -Omics and Fungal Pathogen Outbreaks in Crops
3. -Omics and Fungal Pathogen Outbreaks in Humans
4. -Omics and Fungal Pathogen Outbreaks in Wildlife
5. Discussion
6. Case Studies
6.1. Case Study 1: Magnaporthe Oryzae Genome Reconstruction and Proteomics Yield Insights into Pathogen Biology and Infection Process
6.2. Case Study 2: Genomics to Inform Management Candida Auris Nosociomal Infection Outbreaks in the UK
6.3. Case Study 3: Population Genomics and Phylogenomics to Identify the Causal Agent, Emergence and Dispersal of a Cryptococcosis Outbreak in the Pacific North West
6.4. Case Study 4: Novel -Omics-Based Diagnostics and Point of Origin Investigations for Aspergillus Fumigatus Infection in Kākāpō
6.5. Case Study 5: Investigation into Host Defences against Infection by Batrachochytrium Dendrobatidis (Bd) via a Metabolomics Approach
Author Contributions
Funding
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef]
- Head, M.G.; Fitchett, J.R.; Atun, R.; May, R.C. Systematic analysis of funding awarded for mycology research to institutions in the UK, 1997–2010. BMJ Open 2014, 4, e004129. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Netea, M.G. Medical mycology and fungal immunology: New research perspectives addressing a major world health challenge. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150462. [Google Scholar] [CrossRef] [PubMed]
- Bongomin, F.; Gago, S.; Oladele, R.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases—Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.E.; Park, B.J. Think Fungus—Prevention and Control of Fungal Infections. Emerg. Infect. Dis. 2013, 19, 1688–1689. [Google Scholar] [CrossRef]
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef]
- Benedict, K.; Richardson, M.; Vallabhaneni, S.; Jackson, B.R.; Chiller, T. Emerging issues, challenges, and changing epidemiology of fungal disease outbreaks. Lancet Infect. Dis. 2017, 17, e403–e411. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. In The Fungal Kingdom; ASM Press: Washington, DC, USA, 2017; pp. 79–95. [Google Scholar]
- Conesa, A.; Beck, S. Making multi-omics data accessible to researchers. Sci. Data 2019, 6, 251. [Google Scholar] [CrossRef]
- Seyedmousavi, S.; Guillot, J.; Arné, P.; Arné, A.; Sybren De Hoog, G.; Mouton, J.W.; Melchers, W.J.G.; Verweij, P.E. Aspergillus and aspergilloses in wild and domestic animals: A global health concern with parallels to human disease. Med. Mycol. 2015, 53, 765–797. [Google Scholar] [CrossRef]
- Menardo, F.; Praz, C.R.; Wyder, S.; Ben-David, R.; Bourras, S.; Matsumae, H.; McNally, K.E.; Parlange, F.; Riba, A.; Roffler, S.; et al. Hybridization of powdery mildew strains gives rise to pathogens on novel agricultural crop species. Nat. Genet. 2016, 48, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.S.; Bouguennec, A.; Confais, J.; Morgant, G.; Leroux, P. Evidence of host-range expansion from new powdery mildew (Blumeria graminis) infections of triticale (×Triticosecale) in France. Plant Pathol. 2011, 60, 207–220. [Google Scholar] [CrossRef]
- Singh, R.P.; Singh, P.K.; Rutkoski, J.; Hodson, D.P.; He, X.; Jørgensen, L.N.; Hovmøller, M.S.; Huerta-Espino, J. Disease Impact on Wheat Yield Potential and Prospects of Genetic Control. Annu. Rev. Phytopathol. 2016, 54, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.M.; Churcher, T.S.; Garner, T.W.J.J.; Fisher, M.C. Persistence of the emerging pathogen Batrachochytrium dendrobatidis outside the amphibian host greatly increases the probability of host extinction. Proc. R. Soc. B Biol. Sci. 2008, 275, 329–334. [Google Scholar] [CrossRef]
- Lindner, D.L.; Gargas, A.; Lorch, J.M.; Banik, M.T.; Glaeser, J.; Kunz, T.H.; Blehert, D.S. DNA-based detection of the fungal pathogen Geomyces destructans in soils from bat hibernacula. Mycologia 2011, 103, 241–246. [Google Scholar] [CrossRef]
- Borer, E.T.; Antonovics, J.; Kinkel, L.L.; Hudson, P.J.; Daszak, P.; Ferrari, M.J.; Garrett, K.A.; Parrish, C.R.; Read, A.F.; Rizzo, D.M. Bridging Taxonomic and Disciplinary Divides in Infectious Disease. Ecohealth 2011, 8, 261–267. [Google Scholar] [CrossRef][Green Version]
- Mcdonald, B.A.; Stukenbrock, E.H. Rapid emergence of pathogens in agro-ecosystems: Global threats to agricultural sustainability and food security. Philos. Trans. Biol. Sci. 2016, 371, 20160026. [Google Scholar] [CrossRef]
- World Health Organization. Mycotoxins. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/mycotoxins (accessed on 16 October 2020).
- Godfray, H.C.J.; Mason-D’croz, D.; Robinson, S. Food System Consequences of a Fungal Disease Epidemic in a Major Crop. Philos Trans. R. Soc. Lond B. Biol. Sci. 2016, 371, 20150467. [Google Scholar] [CrossRef]
- Bebber, D.P.; Ramotowski, M.A.T.; Gurr, S.J. Crop pests and pathogens move polewards in a warming world. Nat. Clim. Chang. 2013. [Google Scholar] [CrossRef]
- Fausto, A.; Rodrigues, M.L.; Coelho, C. The still underestimated problem of fungal diseases worldwide. Front. Microbiol. 2019, 10, 1–5. [Google Scholar]
- Hodson, D.P. Shifting boundaries: Challenges for rust monitoring. Euphytica 2011, 179, 93–104. [Google Scholar] [CrossRef]
- Dubin, H.J.; Brennan, J.P. Fighting a “Shifty Enemy”. In Millions Fed: Proven Successes in Agricultural Development; Spielman, D.J., Pandya-Lorch, R., Eds.; International Food Policy Research Institute (ifpri): Washington, DC, USA, 2009; p. 19. [Google Scholar]
- Shank, R. Wheat Stem Rust and Dought Effects on Bale Agricultural Production and Future Prospects. 1994. Available online: https://www.africa.upenn.edu/eue_web/s_rust94.htm (accessed on 16 October 2020).
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Agrios, G. Plant Pathology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 978-0-12-044565-3. [Google Scholar]
- Grünwald, N.J.; McDonald, B.A.; Milgroom, M.G. Population Genomics of Fungal and Oomycete Pathogens. Annu. Rev. Phytopathol. 2016, 54, 323–346. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H.; Bataillon, T.; Dutheil, J.Y.; Hansen, T.T.; Li, R.; Zala, M.; McDonald, B.A.; Wang, J.; Schierup, M.H. The making of a new pathogen: Insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res. 2011, 21, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Croll, D.; Gladieux, P.; Soanes, D.M.; Persoons, A.; Bhattacharjee, P.; Hossain, M.S.; Gupta, D.R.; Rahman, M.M.; Mahboob, M.G.; et al. Emergence of wheat blast in Bangladesh was caused by a South American lineage of Magnaporthe oryzae. BMC Biol. 2016, 14, 84. [Google Scholar] [CrossRef]
- Rouxel, T.; Grandaubert, J.; Hane, J.K.; Hoede, C.; van de Wouw, A.P.; Couloux, A.; Dominguez, V.; Anthouard, V.; Bally, P.; Bourras, S.; et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat. Commun. 2011, 2, 202. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; Bataillon, T. A Population Genomics Perspective on the Emergence and Adaptation of New Plant Pathogens in Agro-Ecosystems. PLoS Pathog. 2012, 8, e1002893. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; Banke, S.; Javan-Nikkhah, M.; McDonald, B.A. Origin and domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric speciation. Mol. Biol. Evol. 2007, 24, 398–411. [Google Scholar] [CrossRef]
- Eschenbrenner, C.J.; Feurtey, A.; Stukenbrock, E.H. Population Genomics of Fungal Plant Pathogens and the Analyses of Rapidly Evolving Genome Compartments; Humana: New York, NY, USA, 2020; pp. 337–355. [Google Scholar]
- Irelan, J.T.; Hagemann, A.T.; Selker, E.U. High frequency repeat-induced point mutation (RIP) is not associated with efficient recombination in Neurospora. Genetics 1994, 138, 1093–1103. [Google Scholar]
- Sánchez-Vallet, A.; Hartmann, F.E.; Marcel, T.C.; Croll, D. Nature’s genetic screens: Using genome-wide association studies for effector discovery. Mol. Plant Pathol. 2018, 19, 3–6. [Google Scholar] [CrossRef]
- Zhong, Z.; Marcel, T.C.; Hartmann, F.E.; Ma, X.; Plissonneau, C.; Zala, M.; Ducasse, A.; Confais, J.; Compain, J.; Lapalu, N.; et al. A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytol. 2017, 214, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Zhang, X.; Liu, F.; Peng, G.; Yu, F.; Fernando, D. Identification of resistance loci in Chinese and Canadian canola/rapeseed varieties against Leptosphaeria maculans based on genome-wide association studies. BMC Genom. 2020, 21, 501. [Google Scholar] [CrossRef] [PubMed]
- Trick, W.E.; Fridkin, S.K.; Edwards, J.R.; Hajjeh, R.A.; Gaynes, R.P. National Nosocomial Infections Surveillance System Hospitals. Secular Trend of Hospital-Acquired Candidemia among Intensive Care Unit Patients in the United States during 1989–1999. Clin. Infect. Dis. 2002, 35, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.J.; Newman, K.R.; Moody, M.; Wharton, R.C.; Wade, J.C. Trichosporonosis in Patients with Neoplastic Disease. Medicine 1986, 65, 268–279. [Google Scholar] [CrossRef] [PubMed]
- GAFFI. The Burden of Fungal Disease: New Evidence to Show the Scale of the Problem across the Globe. 2017. Available online: https://www.gaffi.org/the-burden-of-fungal-disease-new-evidence-to-show-the-scale-of-the-problem-across-the-globe/ (accessed on 30 June 2020).
- Bartlett, K.H.; Kidd, S.E.; Kronstad, J.W. The emergence of Cryptococcus gattii in British Columbia and the Pacific Northwest. Curr. Infect. Dis. Rep. 2008, 10, 58–65. [Google Scholar] [CrossRef]
- Badiee, P.; Zare, M. Consideration of Invasive Fungal Infections in Immunocompetent Hosts. Arch. Clin. Infect. Dis. 2017, 12, 66111. [Google Scholar] [CrossRef]
- Kanamori, H.; Rutala, W.A.; Sickbert-Bennett, E.E.; Weber, D.J. Review of Fungal Outbreaks and Infection Prevention in Healthcare Settings During Construction and Renovation. Health Epidemiol. 2015, 61, 433–444. [Google Scholar] [CrossRef]
- Datta, K.; Bartlett, K.H.; Baer, R.; Byrnes, E.; Galanis, E.; Heitman, J.; Hoang, L.; Leslie, M.J.; MacDougall, L.; Magill, S.S.; et al. Spread of Cryptococcus gattii into Pacific Northwest Region of the United States. Emerg. Infect. Dis. 2009, 15, 1185–1191. [Google Scholar] [CrossRef]
- Springer, D.J.; Billmyre, R.B.; Filler, E.E.; Voelz, K.; Pursall, R.; Mieczkowski, P.A.; Larsen, R.A.; Dietrich, F.S.; May, R.C.; Filler, S.G.; et al. Cryptococcus gattii VGIII Isolates Causing Infections in HIV/AIDS Patients in Southern California: Identification of the Local Environmental Source as Arboreal. PLoS Pathog. 2014, 10, e1004285. [Google Scholar] [CrossRef]
- Hoenen, T. Sequencing of Ebola Virus Genomes Using Nanopore Technology. Bio-Protocol 2016, 6. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Ashton, P.; Calus, S.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris article. Emerg. Microbes Infect. 2018, 7, 43. [Google Scholar] [PubMed]
- Ashikawa, S.; Tarumoto, N.; Imai, K.; Sakai, J.; Kodana, M.; Kawamura, T.; Ikebuchi, K.; Murakami, T.; Mitsutake, K.; Maesaki, S.; et al. Rapid identification of pathogens from positive blood culture bottles with the MinION nanopore sequencer. J. Med. Microbiol. 2018, 67, 1589–1595. [Google Scholar] [CrossRef]
- Stone, N.R.H.; Rhodes, J.; Fisher, M.C.; Mfinanga, S.; Kivuyo, S.; Rugemalila, J.; Segal, E.S.; Needleman, L.; Molloy, S.F.; Kwon-Chung, J.; et al. Dynamic ploidy changes drive fluconazole resistance in human cryptococcal meningitis. J. Clin. Investig. 2019, 129, 999–1014. [Google Scholar] [CrossRef]
- Sionov, E.; Chang, Y.C.; Kwon-Chung, K.J. Azole heteroresistance in Cryptococcus neoformans: Emergence of resistant clones with chromosomal disomy in the mouse brain during fluconazole treatment. Antimicrob. Agents Chemother. 2013, 57, 5127–5130. [Google Scholar] [CrossRef]
- Chang, Y.C.; Khanal Lamichhane, A.; Kwon-Chung, K.J. Cryptococcus neoformans, Unlike Candida albicans, Forms Aneuploid Clones Directly from Uninucleated Cells under Fluconazole Stress. MBio 2018, 9. [Google Scholar] [CrossRef]
- Mondo, S.J.; Kuo, R.C.; Singan, V.R. Fungal Epigenomics: Detection and Analysis; Humana Press: New York, NY, USA, 2018; pp. 155–170. [Google Scholar]
- Blehert, D.S. Fungal Disease and the Developing Story of Bat White-nose Syndrome. PLoS Pathog. 2012, 8, e1002779. [Google Scholar] [CrossRef]
- Scheele, B.C.; Pasmans, F.; Skerratt, L.F.; Berger, L.; Martel, A.; Beukema, W.; Acevedo, A.A.; Burrowes, P.A.; Carvalho, T.; Catenazzi, A.; et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 2019, 363, 1459–1463. [Google Scholar] [CrossRef]
- Alley, M.R.; Gartrell, B.D. Wildlife diseases in New Zealand: Recent findings and future challenges. N. Z. Vet. J. 2019, 67, 1–11. [Google Scholar] [CrossRef]
- Lorch, J.M.; Knowles, S.; Lankton, J.S.; Michell, K.; Edwards, J.L.; Kapfer, J.M.; Staffen, R.A.; Wild, E.R.; Schmidt, K.Z.; Ballmann, A.E.; et al. Snake fungal disease: An emerging threat to wild snakes. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, S.J.; Rieux, A.; Farrer, R.A.; Rosa, G.M.; Waldman, B.; Bataille, A.; Kosch, T.A.; Murray, K.A.; Brankovics, B.; Fumagalli, M.; et al. Recent Asian origin of chytrid fungi causing global amphibian declines. Science 2018, 360, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, E.B.; James, T.Y.; Zamudio, K.R.; Poorten, T.J.; Ilut, D.; Rodriguez, D.; Eastman, J.M.; Richards-Hrdlicka, K.; Joneson, S.; Jenkinson, T.S.; et al. Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data. Proc. Natl. Acad. Sci. USA 2013, 110, 9385–9390. [Google Scholar] [CrossRef] [PubMed]
- Farrer, R.A.; Weinert, L.A.; Bielby, J.; Garner, T.W.J.; Balloux, F.; Clare, F.C.; Bosch, J.; Cunningham, A.A.; Weldon, C.; du Preez, L.H.; et al. Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc. Natl. Acad. Sci. USA 2011, 108, 18732–18736. [Google Scholar] [CrossRef]
- Farrer, R.A.; Martel, A.; Verbrugghe, E.; Abouelleil, A.; Ducatelle, R.; Longcore, J.E.; James, T.Y.; Pasmans, F.; Fisher, M.C.; Cuomo, C.A. Genomic innovations linked to infection strategies across emerging pathogenic chytrid fungi. Nat. Commun. 2017, 8, 14742. [Google Scholar] [CrossRef]
- McDonald, C.A.; Ellison, A.R.; Toledo, L.F.; James, T.Y.; Zamudio, K.R. Gene expression varies within and between enzootic and epizootic lineages of Batrachochytrium dendrobatidis (Bd) in the Americas. Fungal Biol. 2020, 124, 34–43. [Google Scholar] [CrossRef]
- Eskew, E.A.; Shock, B.C.; LaDouceur, E.E.B.; Keel, K.; Miller, M.R.; Foley, J.E.; Todd, B.D. Gene expression differs in susceptible and resistant amphibians exposed to Batrachochytrium dendrobatidis. R. Soc. Open Sci. 2018, 5, 170910. [Google Scholar] [CrossRef]
- Ellison, A.R.; DiRenzo, G.V.; McDonald, C.A.; Lips, K.R.; Zamudio, K.R. First in Vivo Batrachochytrium dendrobatidis Transcriptomes Reveal Mechanisms of Host Exploitation, Host-Specific Gene Expression, and Expressed Genotype Shifts. G3 Genes Genomes Genet. 2017. [Google Scholar] [CrossRef]
- Ellison, A.; Zamudio, K.; Lips, K.R.; Muletz-Wolz, C. Temperature-mediated shifts in salamander transcriptomic responses to the amphibian-killing fungus. Mol. Ecol. 2020, 29, 325–343. [Google Scholar] [CrossRef]
- Madison, J.D.; Berg, E.A.; Abarca, J.G.; Whitfield, S.M.; Gorbatenko, O.; Pinto, A.; Kerby, J.L. Characterization of Batrachochytrium dendrobatidis Inhibiting Bacteria from Amphibian Populations in Costa Rica. Front. Microbiol. 2017, 8, 290. [Google Scholar] [CrossRef]
- Becker, M.H.; Walke, J.B.; Cikanek, S.; Savage, A.E.; Mattheus, N.; Santiago, C.N.; Minbiole, K.P.C.; Harris, R.N.; Belden, L.K.; Gratwicke, B. Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142881. [Google Scholar] [CrossRef] [PubMed]
- Bataille, A.; Lee-Cruz, L.; Tripathi, B.; Waldman, B. Skin Bacterial Community Reorganization Following Metamorphosis of the Fire-Bellied Toad (Bombina orientalis). Microb. Ecol. 2018, 75, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Walke, J.B.; Becker, M.H.; Loftus, S.C.; House, L.L.; Teotonio, T.L.; Minbiole, K.P.C.; Belden, L.K. Community Structure and Function of Amphibian Skin Microbes: An Experiment with Bullfrogs Exposed to a Chytrid Fungus. PLoS ONE 2015, 10, e0139848. [Google Scholar] [CrossRef]
- Rebollar, E.A.; Antwis, R.E.; Becker, M.H.; Belden, L.K.; Bletz, M.C.; Brucker, R.M.; Harrison, X.A.; Hughey, M.C.; Kueneman, J.G.; Loudon, A.H.; et al. Using “omics” and integrated multi-omics approaches to guide probiotic selection to mitigate chytridiomycosis and other emerging infectious diseases. Front. Microbiol. 2016, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Gargas, A.; Trest, M.T.; Christensen, M.; Volk, T.J.; Blehert, D.S. Geomyces destructans sp. nov. associated with bat white-nose syndrome. Mycotaxon 2009, 108, 147–154. [Google Scholar] [CrossRef]
- Clark, R.W.; Marchand, M.N.; Clifford, B.J.; Stechert, R.; Stephens, S. Decline of an isolated timber rattlesnake (Crotalus horridus) population: Interactions between climate change, disease, and loss of genetic diversity. Biol. Conserv. 2011, 144, 886–891. [Google Scholar] [CrossRef]
- Leonardi, M.; Librado, P.; Der Sarkissian, C.; Schubert, M.; Alfarhan, A.H.; Alquraishi, S.A.; Al-Rasheid, K.A.S.; Gamba, C.; Willerslev, E.; Orlando, L. Evolutionary Patterns and Processes: Lessons from Ancient DNA. Syst. Biol. 2016, 66, syw059. [Google Scholar] [CrossRef]
- Andam, C.P.; Worby, C.J.; Chang, Q.; Campana, M.G. Microbial Genomics of Ancient Plagues and Outbreaks. Trends Microbiol. 2016, 24, 978–990. [Google Scholar] [CrossRef]
- Spyrou, M.A.; Keller, M.; Tukhbatova, R.I.; Scheib, C.L.; Nelson, E.A.; Andrades Valtueña, A.; Neumann, G.U.; Walker, D.; Alterauge, A.; Carty, N.; et al. Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes. Nat. Commun. 2019, 10, 4470. [Google Scholar] [CrossRef]
- Wagner, D.M.; Klunk, J.; Harbeck, M.; Devault, A.; Waglechner, N.; Sahl, J.W.; Enk, J.; Birdsell, D.N.; Kuch, M.; Lumibao, C.; et al. Yersinia pestis and the plague of Justinian 541–543 AD: A genomic analysis. Lancet Infect. Dis. 2014, 14, 319–326. [Google Scholar] [CrossRef]
- Rieux, A.; Balloux, F. Inferences from tip-calibrated phylogenies: A review and a practical guide. Mol. Ecol. 2016, 25, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Cooper, A. Using Ancient DNA to Understand Evolutionary and Ecological Processes. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 573–598. [Google Scholar] [CrossRef]
- Mykrä, H.; Tolkkinen, M.; Markkola, A.M.; Pirttilä, A.M.; Muotka, T. Phylogenetic clustering of fungal communities in human-disturbed streams. Ecosphere 2016, 7, e01316. [Google Scholar] [CrossRef]
- Noor, E.; Cherkaoui, S.; Sauer, U. Biological insights through omics data integration. Curr. Opin. Syst. Biol. 2019, 15, 39–47. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14. [Google Scholar] [CrossRef]
- Talbot, N.J. On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea. Annu. Rev. Microbiol. 2003, 57, 177–202. [Google Scholar] [CrossRef]
- Greer, C.A.; Webster, R.K. Occurrence, Distribution, Epidemiology, Cultivar Reaction, and Management of Rice Blast Disease in California. Plant Dis. 2001, 85, 1096–1102. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Ebbole, D.J. Magnaporthe as a Model for Understanding Host-Pathogen Interactions. Annu. Rev. Phytopathol. 2007, 45, 437–456. [Google Scholar] [CrossRef]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.R.; Pan, H.; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef]
- Ding, S.; Zhou, X.; Zhao, X.; Xu, J.R. The PMK1 MAP Kinase Pathway and Infection-Related Morphogenesis. In Advances in Genetics, Genomics and Control of Rice Blast Disease; Springer: Dordrecht, The Netherlands, 2009; pp. 13–21. [Google Scholar]
- Soanes, D.M.; Chakrabarti, A.; Paszkiewicz, K.H.; Dawe, A.L.; Talbot, N.J. Genome-wide Transcriptional Profiling of Appressorium Development by the Rice Blast Fungus Magnaporthe oryzae. PLoS Pathog. 2012, 8, e1002514. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Yu, S.; Kim, S.G.; Kim, H.J.; Kang, S.Y.; Hwang, D.H.; Jang, Y.S.; Kang, K.Y. Proteome analysis of rice blast fungus (Magnaporthe grisea) proteome during appressorium formation. Proteomics 2004, 4, 3579–3587. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Jeong, S.H.; Kim, S.H.; Singh, R.; Lee, J.; Cho, Y.S.; Agrawal, G.K.; Rakwal, R.; Jwa, N.S. Secretome analysis of Magnaporthe oryzae using in vitro systems. Proteomics 2012, 12, 878–900. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Gupta, R.; Min, C.W.; Kwon, S.W.; Wang, Y.; Je, B., II; Kim, Y.J.; Jeon, J.S.; Agrawal, G.K.; Rakwal, R.; et al. Proteomics of Rice—Magnaporthe oryzae Interaction: What Have We Learned So Far? Front. Plant Sci. 2019, 10, 1383. [Google Scholar] [CrossRef]
- Satoh, K.; Makimura, K.; Hasumi, Y.; Nishiyama, Y.; Uchida, K.; Yamaguchi, H. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol. Immunol. 2009, 53, 41–44. [Google Scholar] [CrossRef]
- Spivak, E.S.; Hanson, K.E. Candida auris: An Emerging Fungal Pathoge. J. Clin. Microbiol. 2018, 56, e01588-17. [Google Scholar] [CrossRef]
- David, W.E.; Anna, E.S.; Hilary, M.; Ian, M.; Ruth, M.; Phuong, Q.T.; David, G.; Sophie, G.; Lisa, B.; Marcus, M.; et al. A Candida auris Outbreak and Its Control in an Intensive Care Setting. N. Engl. J. Med. 2018, 14, 1322–1353. [Google Scholar]
- Perfect, J.R.; Bicanic, T. Cryptococcosis diagnosis and treatment: What do we know now. Fungal Genet. Biol. 2015, 78, 49–54. [Google Scholar] [CrossRef]
- Byrnes III, E.J.; Bildfell, R.; Frank, S.A.; Mitchell, T.G.; Marr, K.; Heitman, J. Molecular Evidence that the Vancouver Island Cryptococcus gattii Outbreak has Expanded into the United States Pacific Northwest. J. Infect. Dis. 2009, 199, 1081–1086. [Google Scholar] [CrossRef]
- Kidd, S.E.; Hagen, F.; Tscharke, R.L.; Huynh, M.; Bartlett, K.H.; Fyfe, M.; MacDougall, L.; Boekhout, T.; Kwon-Chung, K.J.; Meyer, W. A Rare Genotype of Cryptococcus gattii Caused the Cryptococcosis Outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. USA 2004, 101, 17258–17263. [Google Scholar] [CrossRef]
- Fraser, J.A.; Giles, S.S.; Wenink, E.C.; Geunes-Boyer, S.G.; Wright, J.R.; Diezmann, S.; Allen, A.; Stajich, J.E.; Dietrich, F.S.; Perfect, J.R.; et al. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 2005, 437, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Gillece, J.D.; Schupp, J.M.; Balajee, S.A.; Harris, J.; Pearson, T.; Yan, Y.; Keim, P.; DeBess, E.; Marsden-Haug, N.; Wohrle, R.; et al. Whole Genome Sequence Analysis of Cryptococcus gattii from the Pacific Northwest Reveals Unexpected Diversity. PLoS ONE 2011, 6, e28550. [Google Scholar] [CrossRef] [PubMed]
- D’souza, C.A.; Kronstad, J.W.; Taylor, G.; Warren, R.; Yuen, M.; Hu, G.; Jung, W.H.; Sham, A.; Kidd, S.E.; Tangen, K.; et al. Genome Variation in Cryptococcus gattii, an Emerging Pathogen of Immunocompetent Hosts. MBio 2011, 2, e00342-10. [Google Scholar] [CrossRef] [PubMed]
- Engelthaler, D.M.; Hicks, N.D.; Gillece, J.D.; Roe, C.C.; Schupp, J.M.; Driebe, E.M.; Gilgado, F.; Carriconde, F.; Trilles, L.; Firacative, C.; et al. Cryptococcus gattii in North American Pacific Northwest: Whole-population genome analysis provides insights into species evolution and dispersal. MBio 2014, 5, e01464-14. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Croll, D.; Li, W.; Mieczkowski, P.; Carter, D.A.; Cuomo, C.A.; Kronstad, J.W.; Heitman, J. Highly recombinant VGII Cryptococcus gattii population develops clonal outbreak clusters through both sexual macroevolution and asexual microevolution. MBio 2014, 5, e01494-14. [Google Scholar] [CrossRef]
- Ma, H.; Hagen, F.; Stekel, D.J.; Johnston, S.A.; Sionov, E.; Falk, R.; Polacheck, I.; Boekhout, T.; May, R.C. The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 12980–12985. [Google Scholar] [CrossRef]
- Byrnes, E.J.; Li, W.; Lewit, Y.; Ma, H.; Voelz, K.; Ren, P.; Carter, D.A.; Chaturvedi, V.; Bildfell, R.J.; May, R.C.; et al. Emergence and Pathogenicity of Highly Virulent Cryptococcus gattii Genotypes in the Northwest United States. PLoS Pathog. 2010, 6, e1000850. [Google Scholar] [CrossRef]
- Farrer, R.A.; Voelz, K.; Henk, D.A.; Johnston, S.A.; Fisher, M.C.; May, R.C.; Cuomo, C.A. Microevolutionary traits and comparative population genomics of the emerging pathogenic fungus Cryptococcus gattii. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- BirdLife International. Strigops habroptila. The IUCN Red List of Threatened Species 2018: e.T22685245A129751169. 2018. Available online: https://dx.doi.org/10.2305/IUCN.UK.2018-2.RLTS.T22685245A129751169.en. (accessed on 16 October 2020).
- New Zealand Department of Conservation. Kākāpō Aspergillosis Outbreak. 2020. Available online: https://www.doc.govt.nz/our-work/kakapo-recovery/what-we-do/kakapo-aspergillosis-outbreak/ (accessed on 15 October 2020).
- Heddergott, C.; Calvo, A.M.; Latgé, J.P. The Volatome of Aspergillus fumigatus. Eukaryot. Cell 2014, 13, 1014–1025. [Google Scholar] [CrossRef]
- Koo, S.; Thomas, H.R.; Daniels, S.D.; Lynch, R.C.; Fortier, S.M.; Shea, M.M.; Rearden, P.; Comolli, J.C.; Baden, L.R.; Marty, F.M. A Breath Fungal Secondary Metabolite Signature to Diagnose Invasive Aspergillosis. Clin. Infect. Dis. 2014, 59, 1733–1740. [Google Scholar] [CrossRef]
- Pappalardo, L.; Hoijemberg, P.; Pelczer, I.; Bailey, T.A. NMR-Metabolomics Study on Falcons Affected by Aspergillosis. Curr. Metab. 2014, 2, 155–161. [Google Scholar] [CrossRef]
- Byrne, A.Q.; Vredenburg, V.T.; Martel, A.; Pasmans, F.; Bell, R.C.; Blackburn, D.C.; Bletz, M.C.; Bosch, J.; Briggs, C.J.; Brown, R.M.; et al. Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation. Proc. Natl. Acad. Sci. USA 2019, 116, 20382–20387. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, T.S.; Betancourt Román, C.M.; Lambertini, C.; Valencia-Aguilar, A.; Rodriguez, D.; Nunes-de-Almeida, C.H.L.L.; Ruggeri, J.; Belasen, A.M.; da Leite Silva, D.; Zamudio, K.R.; et al. Amphibian-killing chytrid in Brazil comprises both locally endemic and globally expanding populations. Mol. Ecol. 2016, 25, 2978–2996. [Google Scholar] [CrossRef] [PubMed]
- Farrer, R.A.; Fisher, M.C. Describing Genomic and Epigenomic Traits Underpinning Emerging Fungal Pathogens. In Advances in Genetics; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 100, pp. 309–328. ISBN 9780128132616. [Google Scholar]
- Rosenblum, E.B.; Stajich, J.E.; Maddox, N.; Eisen, M.B. Global Gene Expression Profiles for Life Stages of the Deadly Amphibian Pathogen Batrachochytrium Dendrobatidis. Proc. Natl. Acad. Sci. USA 2008, 105, 17034–17039. [Google Scholar] [CrossRef] [PubMed]
- Woodhams, D.C.; Ardipradja, K.; Alford, R.A.; Marantelli, G.; Reinert, L.K.; Rollins-Smith, L.A. Resistance to chytridiomycosis varies among amphibian species and is correlated with skin peptide defenses. Anim. Conserv. 2007, 10, 409–417. [Google Scholar] [CrossRef]
- Bell, S.C.; Alford, R.A.; Garland, S.; Padilla, G.; Thomas, A.D. Screening bacterial metabolites for inhibitory effects against Batrachochytrium dendrobatidis using a spectrophotometric assay. Dis. Aquat. Organ. 2013, 103, 77–85. [Google Scholar] [CrossRef]
- Bates, K.A.; Clare, F.C.; O’hanlon, S.; Bosch, J.; Brookes, L.; Hopkins, K.; Mclaughlin, E.J.; Daniel, O.; Garner, T.W.J.; Fisher, M.C.; et al. Amphibian chytridiomycosis outbreak dynamics are linked with host skin bacterial community structure. Nat. Commun. 2018, 9, 693. [Google Scholar] [CrossRef]
- Becker, C.G.; Rodriguez, D.; Longo, A.V.; Toledo, L.F.; Lambertini, C.; Leite, D.S.; Haddad, C.F.B.; Zamudio, K.R. Deforestation, host community structure, and amphibian disease risk. Basic Appl. Ecol. 2016, 17, 72–80. [Google Scholar] [CrossRef]
- Jenke-Kodama, H.; Müller, R.; Dittman, E. Evolutionary mechansims underlying secondary metabolite diversity. In Natural Compounds as Drugs Volume I. Progress in Drug Research; Petersen, F., Amstutz, R., Eds.; Birkhäuser: Basel, Switzerland, 2008. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, P.N.; Brookes, L.M.; Edwards, H.M.; Fisher, M.C.; Jervis, P.; Kappel, D.; Sewell, T.R.; Shelton, J.M.G.; Skelly, E.; Rhodes, J.L. Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections. Life 2020, 10, 315. https://doi.org/10.3390/life10120315
Ghosh PN, Brookes LM, Edwards HM, Fisher MC, Jervis P, Kappel D, Sewell TR, Shelton JMG, Skelly E, Rhodes JL. Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections. Life. 2020; 10(12):315. https://doi.org/10.3390/life10120315
Chicago/Turabian StyleGhosh, Pria N., Lola M. Brookes, Hannah M. Edwards, Matthew C. Fisher, Phillip Jervis, Dana Kappel, Thomas R. Sewell, Jennifer M.G. Shelton, Emily Skelly, and Johanna L. Rhodes. 2020. "Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections" Life 10, no. 12: 315. https://doi.org/10.3390/life10120315
APA StyleGhosh, P. N., Brookes, L. M., Edwards, H. M., Fisher, M. C., Jervis, P., Kappel, D., Sewell, T. R., Shelton, J. M. G., Skelly, E., & Rhodes, J. L. (2020). Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections. Life, 10(12), 315. https://doi.org/10.3390/life10120315